Cardiolipin (CL) is a mitochondrial signature phospholipid that plays a pivotal role in mitochondrial dynamics, membrane structure, oxidative phosphorylation, mtDNA bioenergetics, and mitophagy. The depletion or abnormal acyl composition of CL causes mitochondrial dysfunction, which is implicated in the pathogenesis of aging and age-related disorders. However, the molecular mechanisms by which mitochondrial dysfunction causes age-related diseases remain poorly understood. Studies in the field has identified acyl-CoA: lysocardiolipin acyltransferase 1 (ALCAT1), an acyltransferase upregulated by oxidative stress, as a key enzyme that promotes mitochondrial dysfunction in age-related diseases. ALCAT1 catalyzes CL remodeling with very-long-chain polyunsaturated fatty acids, such as docosahexaenoic acid (DHA). Enrichment of DHA renders CL highly sensitive to oxidative damage by reactive oxygen species (ROS). Oxidized CL becomes a new source of ROS in the form of lipid peroxides, leading to a vicious cycle of oxidative stress, CL depletion, and mitochondrial dysfunction. Consequently, ablation or the pharmacological inhibition of ALCAT1 have been shown to mitigate obesity, type 2 diabetes, heart failure, cardiomyopathy, fatty liver diseases, neurodegenerative diseases, and cancer.
- ALCAT1
- cardiolipin
- aging
- age-related diseases
- mitochondria
- mitochondrial dysfunction
1. Cardiolipin (CL) Remodeling and Acyl Composition
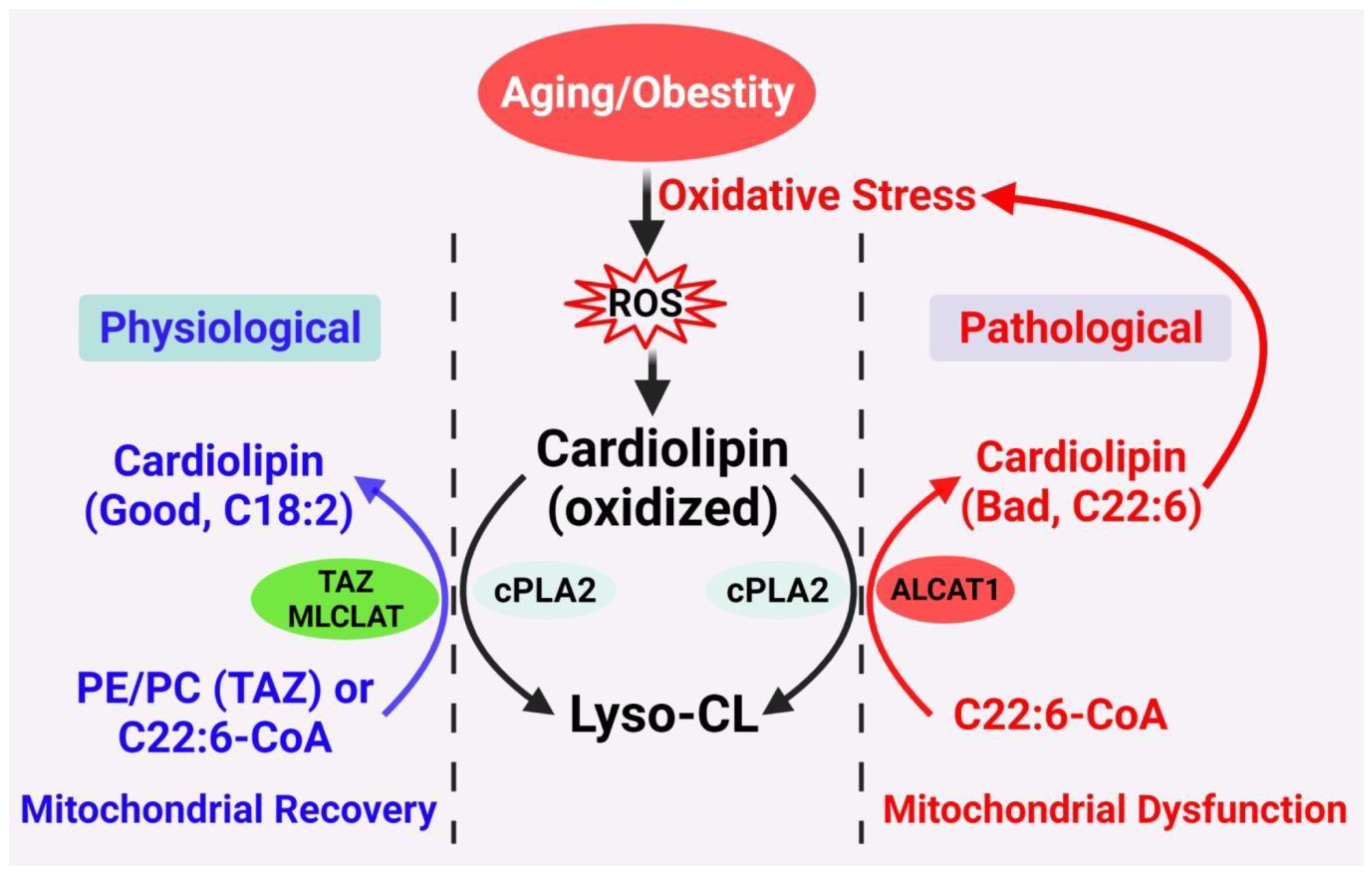
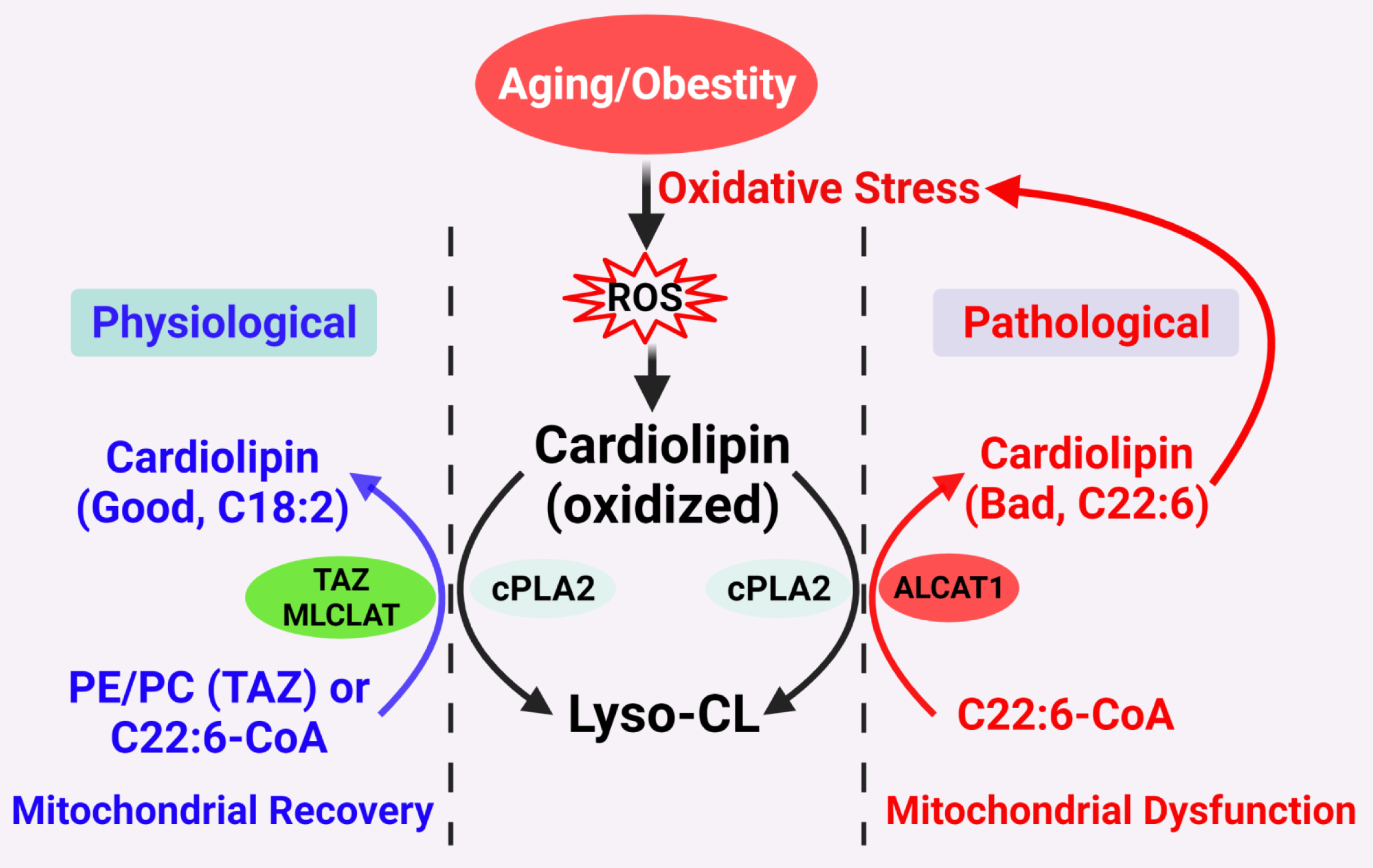 Figure 1. Proposed CL remodeling pathways in obesity and aging. Figure 1. Proposed CL remodeling pathways in obesity and aging. Aging and obesity causes oxidative stress, leading to the production of ROS. CL oxidation by ROS triggers the remodeling of its fatty acyl chains, which begins with hydrolysis of oxidized CL by phospholipase A2 (cPLA2), followed by acylation of lysocardiolipin by either tafazzin (TAZ), MLCLAT, or ALCAT1. TAZ is a transacylase that catalyzes physiological remodeling of CL with other phospholipids, such as PC and PE, whereas MLCLAT catalyzes remodeling of CL with MLCL and linoleoyl-CoA, leading to mitochondrial recovery. ALCAT1 catalyzes remodeling of CL with both MLCL or dilysocardiolipin and docosahexaenoic-CoA (C22:6) as substrates, leading to CL peroxidation by ROS and mitochondrial dysfunction.
Figure 1. Proposed CL remodeling pathways in obesity and aging. Figure 1. Proposed CL remodeling pathways in obesity and aging. Aging and obesity causes oxidative stress, leading to the production of ROS. CL oxidation by ROS triggers the remodeling of its fatty acyl chains, which begins with hydrolysis of oxidized CL by phospholipase A2 (cPLA2), followed by acylation of lysocardiolipin by either tafazzin (TAZ), MLCLAT, or ALCAT1. TAZ is a transacylase that catalyzes physiological remodeling of CL with other phospholipids, such as PC and PE, whereas MLCLAT catalyzes remodeling of CL with MLCL and linoleoyl-CoA, leading to mitochondrial recovery. ALCAT1 catalyzes remodeling of CL with both MLCL or dilysocardiolipin and docosahexaenoic-CoA (C22:6) as substrates, leading to CL peroxidation by ROS and mitochondrial dysfunction.2. Diabetes and Obesity
3. Fatty Liver Disease
4. Heart Diseases
5. Neurological Diseases
6. Bath Syndrome
7. Cancer
8. Conclusion
How aging promotes the development of various age-related diseases is one of the frontiers of biological research. The current evidences support a unified theory on CL remodeling by ALCAT1 as the root cause of age-related diseases by controlling mitochondrial etiology of these disorders (Figure 2). Although much of the evidences accumulated thus far come from studies on mouse models of age-related diseases, the latest development of potent and selective ALCAT1 inhibitors has made it possible to test this theory in human patients in the near future.
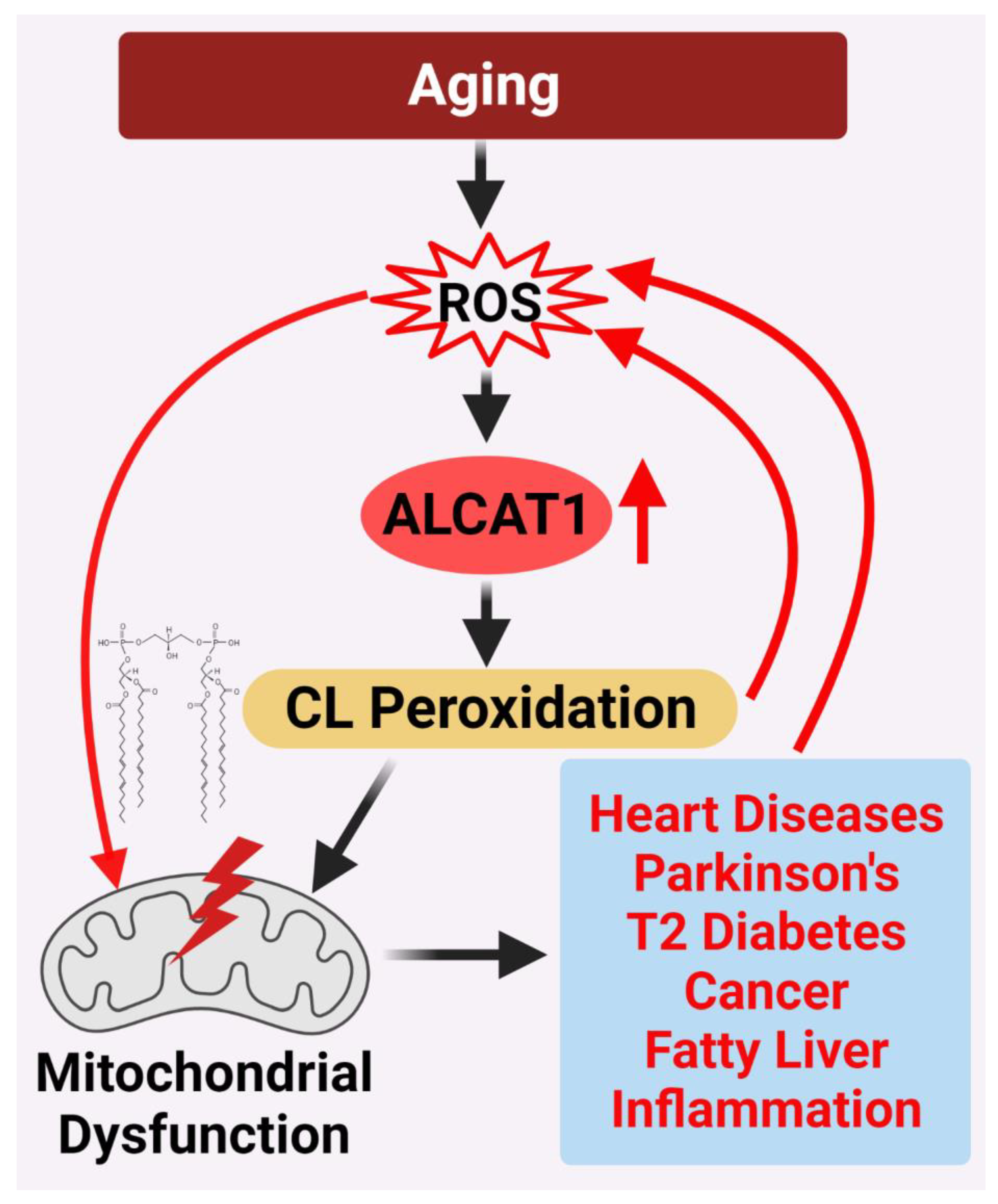 Figure 2. ALCAT1 controls mitochondrial etiology of age-related diseases in response to oxidative stress. ALCAT1 expression is upregulated by ROS from oxidative stress associated with age-related diseases. CL remodeling with PUFA by ALCAT1 causes CL peroxidation, leading to a vicious cycle of ROS production, CL peroxidation, and mitochondrial dysfunction which promote the development of age-related metabolic diseases.
Figure 2. ALCAT1 controls mitochondrial etiology of age-related diseases in response to oxidative stress. ALCAT1 expression is upregulated by ROS from oxidative stress associated with age-related diseases. CL remodeling with PUFA by ALCAT1 causes CL peroxidation, leading to a vicious cycle of ROS production, CL peroxidation, and mitochondrial dysfunction which promote the development of age-related metabolic diseases.
References
- Sparagna, G.C.; Chicco, A.J.; Murphy, R.C.; Bristow, M.R.; Johnson, C.A.; Rees, M.L.; Maxey, M.L.; McCune, S.A.; Moore, R.L. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J. Lipid Res. 2007, 48, 1559–1570.
- Jia, D.; Zhang, J.; Nie, J.; Andersen, J.P.; Rendon, S.; Zheng, Y.; Liu, X.; Tian, Z.; Shi, Y. Cardiolipin Remodeling by ALCAT1 Links Hypoxia to Coronary Artery Disease by Promoting Mitochondrial Dysfunction. Mol. Ther. 2021.
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metabolism. 2010, 12, 154–165.
- Han, X.; Yang, J.; Cheng, H.; Yang, K.; Abendschein, D.R.; Gross, R.W. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 2005, 44, 16684–16694.
- Schlame, M.; Horvath, L.; Vigh, L. Relationship between lipid saturation and lipid-protein interaction in liver mitochondria modified by catalytic hydrogenation with reference to cardiolipin molecular species. Biochem. J. 1990, 265, 79–85.
- Yamaoka-Koseki, S.; Urade, R.; Kito, M. Cardiolipins from rats fed different dietary lipids affect bovine heart cytochrome c oxidase activity. J. Nutr. 1991, 121, 956–958.
- Nomura, K.; Imai, H.; Koumura, T.; Kobayashi, T.; Nakagawa, Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem. J. 2000, 351, 183–193.
- Hostetler, K.Y.; Galesloot, J.M.; Boer, P.; Van Den Bosch, H. Further studies on the formation of cardiolipin and phosphatidylglycerol in rat liver mitochondria. Effect of divalent cations and the fatty acid composition of CDP-diglyceride. Biochim. Biophys. Acta. 1975, 380, 382–389.
- Rustow, B.; Schlame, M.; Rabe, H.; Reichmann, G.; Kunze, D. Species pattern of phosphatidic acid, diacylglycerol, CDP-diacylglycerol and phosphatidylglycerol synthesized de novo in rat liver mitochondria. Biochim. Biophys. Acta. 1989, 1002, 261–263.
- Schlame, M.; Rustow, B.; Kunze, D.; Rabe, H.; Reichmann, G. Phosphatidylglycerol of rat lung. Intracellular sites of formation de novo and acyl species pattern in mitochondria, microsomes and surfactant. Biochem. J. 1986, 240, 247–252.
- Chen, D.; Zhang, X.-Y.; Shi, Y. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem. J. 2006, 398, 169–176.
- Houtkooper, R.H.; Akbari, H.; van Lenthe, H.; Kulik, W.; Wanders, R.J.A.; Frentzen, M.; Vaz, F.M. Identification and characterization of human cardiolipin synthase. FEBS Lett. 2006, 580, 3059–3064.
- Lu, B.; Xu, F.Y.; Jiang, Y.J.; Choy, P.C.; Hatch, G.M.; Grunfeld, C.; Feingold, K.R. Cloning and characterization of a cDNA encoding human cardiolipin synthase (hCLS1). J. Lipid Res. 2006, 47, 1140–1145.
- Shi, Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J. Biomed. Res. 2010, 24, 6–15.
- Mejia, E.M.; Hatch, G.M. Mitochondrial phospholipids: Role in mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 99–112.
- Claypool, S.M.; Koehler, C.M. The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 2012, 37, 32–41.
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288.
- Xu, Y.; Kelley, R.I.; Blanck, T.J.J.; Schlame, M. Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 2003, 278, 51380–51385.
- Cao, J.; Liu, Y.; Lockwood, J.; Burn, P.; Shi, Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 2004, 279, 31727–31734.
- Taylor, W.A.; Hatch, G.M. Purification and characterization of monolysocardiolipin acyltransferase from pig liver mitochondria. J. Biol. Chem. 2003, 278, 12716–12721.
- Taylor, W.A.; Hatch, G.M. Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1). J. Biol. Chem. 2009, 284, 30360–30371.
- Taylor, W.A.; Mejia, E.M.; Mitchell, R.W.; Choy, P.C.; Sparagna, G.C.; Hatch, G.M. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS ONE 2012, 7, e48628.
- Liu, X.; Ye, B.; Miller, S.; Yuan, H.; Zhang, H.; Tian, L.; Nie, J.; Imae, R.; Arai, H.; Li, Y.; et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell Biol. 2012, 32, 4493–4504.
- Song, C.; Zhang, J.; Qi, S.; Liu, Z.; Zhang, X.; Zheng, Y.; Andersen, J.P.; Zhang, W.; Strong, R.; Martinez, P.A.; et al. Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson’s diseases. Aging Cell 2019, 18, e12941.
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496.
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898.
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.-A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800.
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761.
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948.
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622.
- Li, J.; Liu, X.; Wang, H.; Zhang, W.; Chan, D.C.; Shi, Y. Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc. Natl. Acad. Sci. USA 2012, 109, 6975–6980.
- Ziaeian, B.; Fonarow, G.C. Epidemiology and Etiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378.
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724.
- Tocchi, A.; Quarles, E.K.; Basisty, N.; Gitari, L.; Rabinovitch, P.S. Mitochondrial dysfunction in cardiac aging. Biochim. Biophys. Acta 2015, 1847, 1424–1433.
- Pangborn, M.C. Isolation and purification of a serologically active phospholipid from beef heart. J. Biol. Chem. 1942, 143, 247–256.
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44.
- Hsu, P.; Shi, Y. Regulation of autophagy by mitochondrial phospholipids in health and diseases. Biochim. Biophys. Acta 2016.
- Lee, H.-J.; Mayette, J.; Rapoport, S.I.; Bazinet, R.P. Selective remodeling of cardiolipin fatty acids in the aged rat heart. Lipids Health Dis. 2006, 5, 2.
- Watkins, S.M.; Carter, L.C.; German, J.B. Docosahexaenoic acid accumulates in cardiolipin and enhances HT-29 cell oxidant production. J. Lipid Res. 1998, 39, 1583–1588.
- Ng, Y.; Barhoumi, R.; Tjalkens, R.B.; Fan, Y.Y.; Kolar, S.; Wang, N.; Lupton, J.R.; Chapkin, R.S. The role of docosahexaenoic acid in mediating mitochondrial membrane lipid oxidation and apoptosis in colonocytes. Carcinogenesis 2005, 26, 1914–1921.
- Han, X.; Yang, J.; Yang, K.; Zhao, Z.; Abendschein, D.R.; Gross, R.W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: A shotgun lipidomics study. Biochemistry 2007, 46, 6417–6428.
- Sparagna, G.C.; Lesnefsky, E.J. Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 2009, 53, 290–301.
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic. Biol. Med. 2010, 48, 1286–1295.
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 2009, 45, 643–650.
- Wang, W.; Ni, L.; Yu, Q.; Xiong, J.; Liu, H.C.; Rosenwaks, Z. Expression of the Lycat gene in the mouse cardiovascular and female reproductive systems. Dev. Dyn. 2010, 239, 1827–1837.
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30.
- Abou-Sleiman, P.M.; Muqit, M.M.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006, 7, 207–219.
- Agrawal, I.; Jha, S. Mitochondrial Dysfunction and Alzheimer’s Disease: Role of Microglia. Front. Aging Neurosci. 2020, 12.
- Kiebish, M.A.; Han, X.; Cheng, H.; Chuang, J.H.; Seyfried, T.N. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: Lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 2008, 49, 2545–2556.
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barcelo-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell Biol. 2005, 25, 10190–10201.
- Bayir, H.; Tyurin, V.A.; Tyurina, Y.Y.; Viner, R.; Ritov, V.B.; Amoscato, A.A.; Zhao, Q.; Zhang, X.J.; Janesko-Feldman, K.L.; Alexander, H.; et al. Selective early cardiolipin peroxidation after traumatic brain injury: An oxidative lipidomics analysis. Ann. Neurol. 2007, 62, 154–169.
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat. Commun. 2018, 9, 817.
- Barth, P.G.; Valianpour, F.; Bowen, V.M.; Lam, J.; Duran, M.; Vaz, F.M.; Wanders, R.J. X- linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update. Am. J. Med. Genet. Part. A 2004, 126, 349–354.
- Neuwald, A.F. Barth syndrome may be due to an acyltransferase deficiency. Curr. Biol. 1997, 7, R465–R466.
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; Van der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.; Van ‘t Veer-Korthof, E.T.; Van der Harten, J.J.; Sobotka-Plojhar, M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355.
- Vreken, P.; Valianpour, F.; Nijtmans, L.G.; Grivell, L.A.; Plecko, B.; Wanders, R.J.; Barth, P.G. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 2000, 279, 378–382.
- Schlame, M.; Towbin, J.A.; Heerdt, P.M.; Jehle, R.; DiMauro, S.; Blanck, T.J. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 2002, 51, 634–637.
- Bissler, J.J.; Tsoras, M.; Goring, H.H.; Hug, P.; Chuck, G.; Tombragel, E.; McGraw, C.; Schlotman, J.; Ralston, M.A.; Hug, G. Infantile dilated X-linked cardiomyopathy, G4.5 mutations, altered lipids, and ultrastructural malformations of mitochondria in heart, liver, and skeletal muscle. Lab. Investig. 2002, 82, 335–344.
- Zhang, J.; Liu, X.; Nie, J.; Shi, Y. Restoration of mitophagy ameliorates cardiomyopathy in Barth syndrome. Autophagy 2022, 1–16.
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106.
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129.
- Ahmadpour, S.T.; Mahéo, K.; Servais, S.; Brisson, L.; Dumas, J.-F. Cardiolipin, the Mitochondrial Signature Lipid: Implication in Cancer. Int. J. Mol. Sci. 2020, 21, 8031.
- Zhong, H.; Xiao, M.; Zarkovic, K.; Zhu, M.; Sa, R.; Lu, J.; Tao, Y.; Chen, Q.; Xia, L.; Cheng, S.; et al. Mitochondrial control of apoptosis through modulation of cardiolipin oxidation in hepatocellular carcinoma: A novel link between oxidative stress and cancer. Free. Radic. Biol. Med. 2017, 102, 67–76.
- Feng, H.M.; Zhao, Y.; Zhang, J.P.; Zhang, J.H.; Jiang, P.; Li, B.; Wang, C. Expression and potential mechanism of metabolism-related genes and CRLS1 in non-small cell lung cancer. Oncol. Lett. 2018, 15, 2661–2668.
- Pathak, S.; Meng, W.J.; Zhang, H.; Gnosa, S.; Nandy, S.K.; Adell, G.; Holmlund, B.; Sun, X.F. Tafazzin protein expression is associated with tumorigenesis and radiation response in rectal cancer: A study of Swedish clinical trial on preoperative radiotherapy. PLoS ONE 2014, 9, e98317.
- Li, X.; Wu, M.; An, D.; Yuan, H.; Li, Z.; Song, Y.; Liu, Z. Suppression of Tafazzin promotes thyroid cancer apoptosis via activating the JNK signaling pathway and enhancing INF2- mediated mitochondrial fission. J. Cell Physiol. 2019.
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803.
- Xiong, J.-W.; Yu, Q.; Zhang, J.; Mably, J.D. An acyltransferase controls the generation of hematopoietic and endothelial lineages in zebrafish. Circ. Res. 2008, 102, 1057–1064.
- Huang, L.S.; Kotha, S.R.; Avasarala, S.; VanScoyk, M.; Winn, R.A.; Pennathur, A.; Yashaswini, P.S.; Bandela, M.; Salgia, R.; Tyurina, Y.Y.; et al. Lysocardiolipin acyltransferase regulates NSCLC cell proliferation and migration by modulating mitochondrial dynamics. J. Biol. Chem. 2020, 295, 13393–13406.