In the pace of time, numerous platforms for rapid nucleic acid (NA) detection have been developed; however, they may not be able to satisfy the ASSURED (Affordable, Sensitive, Specific, User-friendly, Rapid and Robust, Equipment-free and Deliverable to end-users) parameters set by the World Health of Organization at the same time. ll rRecent studies have established that the CRISPR/Cas system possesses the potential to fulfil ASSURED criteria and demonstrated that it may be repurposed for the detection of contaminants, enzymes, proteins, analytes, and plant diseases, rather than being limited to clinical settings and nucleic acids. Several platforms have yet to be translated into clinical applications and must be brought into clinical settings from the laboratory.
1. CRISPR-Cas Endonuclease System: An Overview
The existence of CRISPR-associated nuclease (Cas) system was discovered in
Escherichia coli by Japanese researchers in the late 1980s [
5,
8]. Subsequently, the CRISPR/Cas systems were also reported and recognized as acquired immunity systems from other bacterial strains and halophilic archaea [
9,
10,
11]. These represent an unusual repetitive DNA sequence, and consequently were termed CRISPR by Jansen et al., in 2002 [
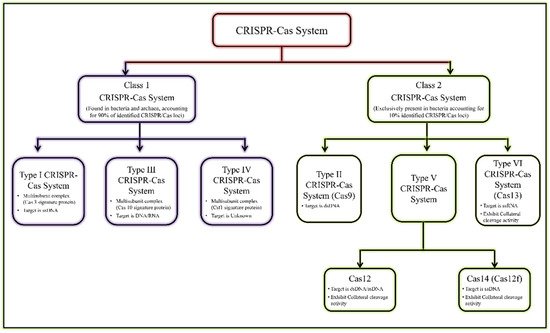
12]. The recent classification of the CRISPR/Cas system is given in
Figure 1 [
13,
14].
Figure 1. General classification of CRISPR/Cas system.
The most reliable and validated genome editing tools are type II, type V, and type VI CRISPR effectors [
14,
15,
16,
17]. Cas recognises and cleaves target sequences in DNA/RNA with the help of guide RNA (gRNA) [
3]. Jennifer Doudna and Emmanuelle Charpentier (2020 Nobel Prize laureate) discovered CRISPR–Cas9 gene-editing tools in 2012 [
18]. Their breakthrough redefined the CRISPR/Cas system for genome editing [
18,
19]. CRISPR–Cas9 has also been employed for genetic marker, typing, and epidemiological purposes, as an antimicrobial agent against pathogenic bacteria [
16,
19,
20,
21]. In addition to developing model cell lines and transgenic animals/plants, the CRISPR/Cas9 system’s flexibility allows it to be employed in understanding disease mechanisms, disease targets, and for transcriptional control [
21]. Beyond these uses, Cas9 unique cleavage activity has also been exploited to construct ultrasensitive DNA biosensing platforms [
3,
22].
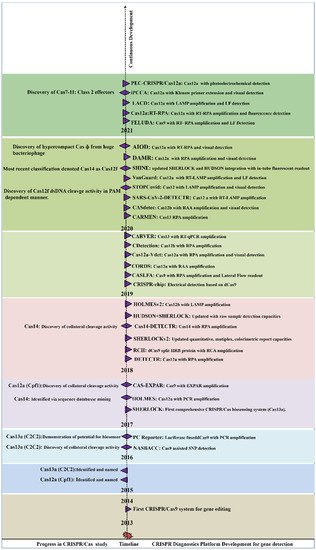
After 2014, many endonucleases were discovered, including Cas12b, Cas13a, CasRx, and Cas14 [
16,
23,
24,
25]. Single crRNA-guided cis (target nucleic acid) and trans (non-target nucleic acid) cleavage activity of Cas12a and Cas13a were discovered in the following years [
26,
27,
28]. Recently, hypercompact CasΦ and Cas7-11 CRISPR/Cas systems were discovered. The CasΦ acts as a DNA cutter which was uniquely identified from genomes of huge bacteriophages and is half the weight of Cas9 and Cas12 [
29], whereas Cas7-11 a new CRISPR Class 1 effector, originated from the fusion of Cas11 and Cas7 units derived from CRISPR subtype III-D. Similarly to Class 2 effectors, Cas7-11 processes crRNA and targets the RNA:Spacer duplex without any collateral activity [
29,
30]. A new perspective for using CRISPR-based diagnostics was promoted following the discovery of trans-cleavage activity (collateral cleavage activity) towards NA. The collateral cleavage of the ssRNA or ssDNA reporter results in a fluorescent signal as a readout, upon the activation of molecular sensors. By utilizing the collateral cleavage activity, several diagnostic platforms were developed including SHERLOCK, DETECTR, CARP, HOLMES [
28,
31,
32].
Figure 2 summarizes the periodic discovery and development of diagnostic platforms.
Figure 2. Year-wise Discovery and development of CRISPR/Cas based Nucleic acid detection platform.
2. Mechanism of CRISPR/Cas Based Detection
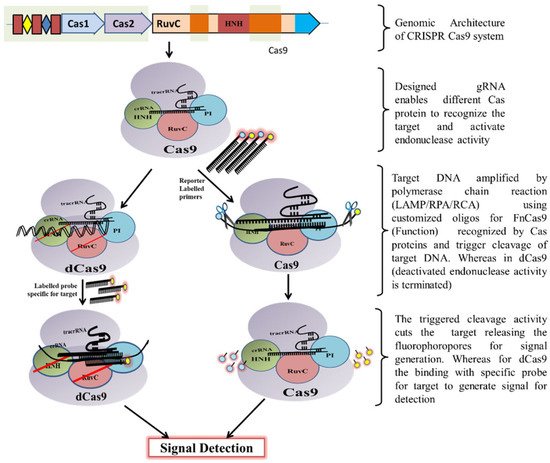
Classically, the CRISPR-Cas9 system is utilized by bacteria to destruct foreign DNA that enters the bacteria through horizontal gene transfer. Initially, a unique non-coding RNA trans-activating CRISPR RNA (tracrRNA) (transcribed separately) hybridizes to the precursor CRISPR transcript (pre-crRNA) through repeat sequences for post-transcriptional processing and endonucleolytic cleavage by RNaseIII to generate the CRISPR RNAs (crRNA) and forms a dual RNA hybrid structure [
33]. This dual RNA guide directs Cas9 for dsDNA cleavage complementary to the target sequence located adjacent to PAM and cleaves each strand with distinct nucleases (HNH or RuvC) (
Figure 3). Engineered sgRNA that combines crRNA and tracrRNA can simplify the system. The protospacer adjacent motif (PAM) sequence is NGG for CRISPR/Cas9, which occurs once in every 8 bps or 4 bps of random DNA, enables the design of sgRNA more conveniently [
34]. An in vitro double-strand break (DSB) of target DNA caused by the CRISPR/Cas9 foreign DNA destruction programme was a breakthrough and created the basis for its gene-editing toolbox application [
35]. This characteristic of Cas9 activation upon target recognition via gRNA possesses great potential in genotyping and gene detection. The ‘magic power’ of Cas9 in biosensing applications has already been established [
34,
36,
37,
38].
Figure 3. Schematic diagram showing CRISPR/Cas9 mechanism for DNA detection.
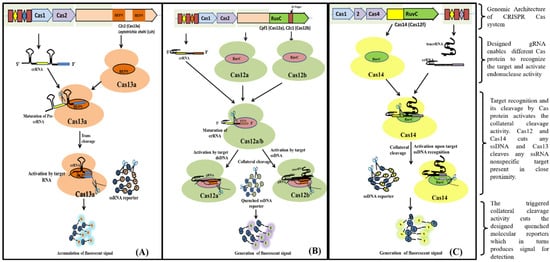
Cas13 nucleases target ssRNA and are a Type VI CRISPR-Cas signature protein. The exclusion of the tracrRNA requirement is the distinctive feature of Cas13 effectors (HEPN)-binding domains (
Figure 4A). Cas13a has been shown to possess dual RNAse activity. First RNAse activity leads to crRNA maturation, while second RNAse activity is responsible for RNA-activated single-stranded RNA destruction [
39,
40]. The presence of dual RNase activity of Cas13 is due to the presence of two RNase catalytic pockets, namely the REC lobe (containing 1-helical domain) which cleaves pre-crRNA and the NUC lobe (containing two HEPN domain) that recognize and cleave target RNA [
40,
41]. The mechanism of cleavage (target (cis) and non-target RNA (trans) lies in the recognition and binding of Cas13 to a protospacer flanking site (PFS) adjacent to the complementary spacer on the 5’ and 3’ ends of the protospacer for Cas13b nuclease, whereas for Cas13a it is present on the 3’ end of ssRNA. The Cas13 endonuclease enzyme with crRNA induces blunt-end breakage and degrades arbitrarily non-target ssRNA present nearby [
42]. A recent report demonstrated the dependence of cleavage activity on conserved catalytic residues in the two HEPN domains as well as the preference of uracil residue for cleavage [
39].
Figure 4. Schematic diagram showing various steps from Cas endonuclease formation to activation of its collateral cleavage activity. (A) Mechanism of CRISPR/Cas13 cis-activation upon target RNA detection activating it for collateral-cleavage of non-target ssRNA. (B) Mechanism of cis-activation of CRISPR/Cas12a and CRISPR/Cas12b by target dsDNA and ssDNA respectively. Both CRISPR/Cas12a and Cas12b trans-cleave non-target ssDNA upon cis-activation. (C) Mechanism of CRISPR/Cas14 (Cas12f) cis-cleavage activation by target ssDNA detection and trans-cleavage activation for non-target ssDNA upon cis-activation.
Cas12, the type V CRISPR-Cas protein, has a single RNA-guided RuvC domain. The Cas12 effectors are a close relative of Cas9, with a single RuvC nuclease domain, enabling Cas12 to introduce staggered dsDNA breaks [
25]. Cas12a and Cas12b of the Cas12 family are widely studied. Cas12b, but not Cas12a, requires tracrRNA for catalysis, whereas nuclease activity in both of them is activated by a PAM sequence upstream of its target. After crRNA-target binding to protospacers juxtaposed to the PAM consensus, Cas12 RuvC-like nucleases cleave the non-target strand within the protospacer 18 nucleotide (nt) from the PAM using a two-metal ion (
Figure 4B) [
43]. The crRNA/Cas12 complex cleaves dsDNA in a staggered fashion, leaving 5- to 7- nt overhangs [
44,
45,
46].
Cas14 (Cas12f) is a type V CRISPR/Cas system that evolved from a superphylum of symbiotic archaea and was discovered to be a distant relative of Cas12 [
16,
47]. Cas14 targets ssDNA with trans ssDNA cleavage. Harrington et al., discovered that Cas14 evolved independently as the smallest protein (400–700 amino acids) in comparison to other Cas endonucleases (950–1400 amino acids) and that it clusters into the following three subgroups: Cas14a (Cas12f1), Cas14b (Cas12f2), and Cas14c (Cas12f3) [
16,
48]. Cas14 was also called a miniature CRISPR system due to its size. Cas14 is a single effector molecule and has been found to destruct DNA independently without requiring PAM (
Figure 4C) [
16,
42,
49]. Karvelis et al., on the other hand, demonstrated that a T-rich PAM sequence can induce the Cas14 endonuclease to cleave dsDNA with staggered ends [
48,
50]. Although Cas14 possesses conserved functional RuvC nuclease, it lacks the RNAse III enzyme, which processes crRNAs and tracrRNAs, as validated by a biochemical study [
16].
Likewise, the newly discovered bacteriophage-derived CasΦ endonuclease is another hypercompact (70 to 80 kDa) endonuclease with a single RuvC active site for both pre-crRNA processing and DNA cleavage. CasΦ is bigger than Cas14 (40- to 70-kDa) but half the size of other CRISPR-Cas systems (100- to 200-kDa). Unlike Cas14, CasΦ requires T-rich PAM sequences (5’-TBN-3’). For the activation of CasΦ endonuclease activity, a cognate PAM and a 14 nt spacer are required. Similar to type V CRISPR-Cas endonucleases, CasΦ generates staggered 5’-overhangs of 8 to 12 nt. Furthermore, when activated by cis recognition, CasΦ efficiently showed non-specific cleavage activity towards dsDNA and ssDNA within the RuvC active site [
29].