Traditional cell cultures are performed in two-dimensional (2D) systems such as Petri dishes, multiwell plates or flasks. However, they cannot realistically mimic the morphophysiological complexity of the original three-dimensional (3D) in vivo environment from which the cells of specific lines originate. Without opposing animal experimentation but promoting its responsible application, the development of alternative cell culture systems tries to ensure compliance with the 3R principles. Reduction (reduction in the animals used for in vivo tests), Refinement (experimental design optimization to limit stress and affliction to laboratory animals) and Replacement (total or partial replacement of animal testing with alternative valid methods) are increasingly desired and strongly recommended as fundamental ethical aspects in the use of animals in scientific experiments.
- 3D cell cultures
- microfluidics
- lab on chip
1. Introduction
2. Physiological Exchange of Substances
In physiological conditions[28, the exchange of substances and gases between cells and the environment takes place thanks to blood microcirculation at the level of the capillaries. Blood circulates from the arterioles to capillaries29, then to venules and the topology of these vessels changes according to the different tissues that are sprinkled. Some beds are structured as trees30, others as arcades or sinuses or portal systems [32]31]. The capillary density (CD) depends on the varying oxygen and nutrients requirements to keep a stable metabolism. The average CD in human tissue is around 600 per mm3 and it changes according to the different organism’s tissues. The CD is higher in the brain, kidneys, liver and myocardium (around 2500–3000 per mm3), reduced in the phasic units of the skeletal musculature (around 300–400 per mm3) and even lower in the bones, fat, connective tissues and in the tonic units of the skeletal musculature (less than 100 per mm3) [33]. Considering an average capillary diameter of 8 μm and length of 5 mm [34], it can be calculated the average distance between adjacent capillaries which is around 30–40 μm (around 1–3 cell width). To reach a particular cell, molecules exit the capillary and cross one or two cells to reach the target one. A capillary vessel can be considered as a tube consisting of a single endothelial cells’ layer less than 1 μm thick [35]. There are three types of capillaries: (i) the continuous type with cells tightly joined together, which are present in muscles, nerves, and connective tissues; (ii) the fenestrated type, with cells so thin that internal vesicles form small pores 100 nm thick and 6 nm in diameter (typically around 1000 pores/μm2); (iii) the discontinuous type with distinct intercellular gaps (around 5 μm in diameter) and a broken basement membrane, commonly found in organs such as the liver, spleen, and bone marrow, the functions of which include the injection or extraction of whole cells, large molecules and extraneous particles in/from the blood stream [36]. The nutritive and waste substances pass the capillary pores by means of a dynamic equilibrium established between the hydraulic pressure and the osmotic pressure gradients between the blood inside the capillaries and the interstitial fluid in the ECM. In particular, the blood’s osmotic pressure (oncotic pressure) is around 25–30 mmHg and it is higher than the one of interstitial fluid which is around 0 mmHg. The osmotic pressure gradient is constant between the blood circuit and the surrounding tissues including the arterial capillaries and the venous capillaries. While the hydraulic pressure in blood decreases, going from the arterial capillaries (where it is around 40 mmHg) to the venous capillaries (which is around 15 mmHg), in the interstitial fluid it is around 2 mmHg. Since, in the arterial capillaries, the hydraulic pressure in the blood is higher than the oncotic pressure, filtration, a flow that goes from capillaries to tissues, occurs. On the contrary, in the venous capillaries, the oncotic pressure is higher than the hydraulic one and liquids are reabsorbed in capillaries due to a flow from tissues to capillaries (Figure 1).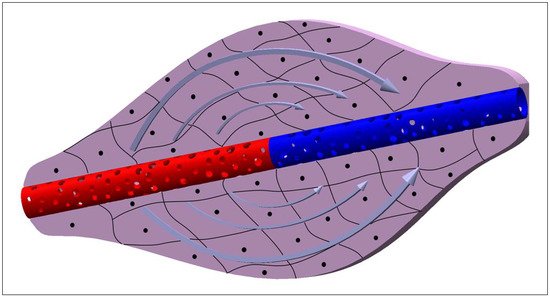
3. Theory behind the Molecule Transport Mechanisms
Referring to the theory behind the movement of particles across a capillary’s membrane, it can be considered a unidimensional motion, assuming the concentration gradient across the membrane as constant. This approximation is certainly valid in the dynamic environment of the biological systems where, while cells consume nutrients and produce wastes, capillaries provide nutrients and remove wastes, keeping the concentration gradients across capillary’s membrane constant. The flux of molecules due to diffusion can be calculated as:- ΔC=(C2−C1) is the concentration gradient of a generic molecule between the external and internal part of the capillary membrane;
- P is the permeability coefficient and can be calculated as:
- Δx is the capillary membrane thickness;
- α is the partition coefficient and can be calculated as:
- N is the number of pores;
- A is the capillary surface;
- R is the pore radius;
- n is the pore density;
- DM is the membrane diffusion coefficient and can be calculated as:
-
ϵ is the hindrance coefficient and it depends on the particle and membrane pore dimension and the trajectory of the particle within the pore and can be calculated as:
- ϵ2 is a coefficient that depends on the trajectory of the particle inside the pore;
- r is the particle radius (it is an approximation which considers the molecules passing the pore to have a spherical shape);
- D is the diffusion coefficient which can be calculated as:
- k is the Boltzman constant;
- T is the temperature;
- η is the blood viscosity;
- Δp=(p2−p1) is the hydraulic pressure gradient across the capillary membrane;
- Δπ=(π2−π1) is the osmotic pressure gradient across the capillary membrane;
- Lp is the filtration coefficient and can be calculated as:
4. Cell Microenvironment: Static and 3D Cell Screening
Mimicking the best possible cellular microenvironment does not only mean having control overflows, since many parameters such as shear stress, cell interactions, pH, CO2, temperature, and O2 variations affect its regulation and balance. Although it is well-known that in any kind of cell screening applications, it is very important to control the cell microenvironment, the current in vitro systems are still far from having an appreciable level of control on it [38]. Generally, supports such as Petri dishes, flasks and vials are used to culture cells in a static condition, leading to temperature and chemical gradients that could make it difficult to maintain homeostasis [39]. In addition, the use of standard static cell culture supports requires a lot of manual procedures, such as the addition of fresh culture medium and the removal of the old one, resulting in time-demanding procedures for the operator and stressful conditions for cells. One of the alternatives to static cell culture procedures is the use of in vivo experiments that are undoubtedly able to reduce the gap between in vitro and in vivo screening procedures. Unfortunately, in vivo experimentation in basic and pre-clinical practice involves a considerable waste of resources, both in monetary and ethical terms, considering the number of animals to be sacrificed. Over the years and with the progress in biomedical and technological fields, there has been a tendency to drastically reduce in vivo experiments using the advanced alternatives to animal testing towards the 3Rs (Replacement, Reduction, Refinement) approach. [40][41][42]. Although replacing should be the main purpose of the 3Rs, its implementation in the short-term is ambitious, while minimizing the number of animals and refining their welfare should be feasible in the short/middle-term [43]. A solid alternative to animal tests is cell scaffolds, as 3D cell culture can effectively mimic the cellular and tissue microarchitecture [44][45]. Both for pharmacological screening and pathologies modelling, 3D scaffolds represent one of the most successful platforms for biomedical applications [46][47][48][49]. Dattola et al. developed a poly(vinyl) alcohol (PVA) 3D scaffold where stem cells grew and differentiated into cardiac cells (Figure 2) [50]. These scaffolds mimicked the mechanical properties of ECM in which cardiomyocytes proliferated in vivo, demonstrated by the contractile property detected in the cardiomyocytes grown on the proposed scaffold. However, it was found that cells colonized only the outermost part of the scaffold, since they could not survive deep into the bulk volume, because the nutrients were not properly provided in the innermost layers of the 3D scaffolds.
| Microfluidic Platform Type | Application | Cell Lines | References |
|---|---|---|---|
| Resin 3D-printed system (VeroClear, MED610 resins) |
Cell Culture, LC-MS/MS single cell analysis | BPAECs (Bovine Pulmonary Artery Endothelial Cells), MDCK (Madin-Darby Canine Kidney) | [52] |
| Microwell-based PDMS-membrane-PDMS sandwich multilayer chips | Spheroid formation, OoC | C3A (liver) | [53] |
| Two-stage temperature-controlling system used to generate decellularized extracellular matrix (dECM) hydrogel microspheres | dECM hydrogels microsphere formation, cell culture | Schwann cells (nervous tissue), PC12 (adrenal gland) | [54] |
| Injection-molded Polystyrene array |
OoC, angiogenesis | HUVEC (Human Umbilical Vein Endothelial Cells), fibroblasts | [55] |
| PDMS-gut-on-a-chip device either with a straight channel or a non-linear convoluted channel, transwell-embedded hybrid chip | OoC | Caco-2 (colon) | [56] |
| Cyclo-olefin-polymer (COP) transparent bioreactor |
On-chip platelet production | imMKCLs (immortalized MegaKaryocyte progenitor Cell Lines) | [57] |
| PDMS soft lithography replicas of superficial channels 3D-printed in different resins (Clear, Model, Tough, Amber, Dental resins) | OoC | HUVEC (Human Umbilical Vein Endothelial Cells), fibroblasts | [58] |
| PDMS bone-mimicking extracellular matrix composite device | Angiogenesis, OoC | SW620 (colon), MKN74 (stomach) | [59] |
| Single-chamber commercial microfluidic device |
OoC, disease model, drug screening | Primary human hepatocytes, EA.hy926 (human endothelial), U937 (pleural effusion), LX-2 (hepatic stellate cell) | [60] |
| Collagen scaffold | OoC | Caco-2 (colon) | [61] |
| Cellulose-based device | Chemotaxis, invasion assay | A549 (lung) | [62] |
| Polymerized High Internal Phase Emulsion (polyHIPE) system | OoC | hES-MPs (human Embrionic Stem cell-derived Mesenchymal Progenitor cells) | [17] |
| OrganoPlate LiverTox™ | Drug screening, OoC | Induced pluripotent stem cell (iPSC)-derived hepatocytes (iHep), endothelial cells, THP-1 monoblast (peripheral blood) | [63] |
| Injection-molded Polystyrene array |
Drug screening | HeLa (uterus, cervix), NK-92 (peripheral blood) | [64] |
| Resin 3D-printed system (VeroClear) |
Spheroid formation | OSCC (Oral Squamous Cell Carcinoma), HepG2 (liver) | [65] |
| 3D-printed device | Circulating Tumour Cells (CTCs) isolation | MCF-7 (breast), SW480 (colon), PC3 (prostate), 293T (kidney) | [66] |
| PDMS-based device | Spheroid formation, disease model, drug screening, OoC | Rat primary hepatocytes, HSCs (Hepatic Stellate Cells) | [67] |
| PDMS-glass chip and Polycarbonate cover-plates |
Four OoC | EpiIntestinal™, HepaRG (liver), HHStec (Human primary Hepatic Stellate cells), RPTEC/TERT-1 (human proximal tubule) | [68] |
| PDMS-based device | OoC | Hepatocytes from primary and iPS-derived cells | [69] |
| Three-layered glass device | OoC, disease model, drug screening | Primary human hepatocytes, LSECs (Liver Sinusoidal Endothelial Cells), Kupffer cells (liver) | [70] |
| Three-layered glass device | OoC, disease model, drug screening | Primary human hepatocytes, iPSC (induced-Pluripotent Stem Cells), endothelial cells, Kupffer cells (liver) | [71] |
| Silicon scaffold fabricated by deep reactive ion etching | OoC, disease model, drug screening | PHH (Primary Human Hepatocyte), non-parenchymal cells | [72] |
| PDMS “open-top” device | Angiogenesis, spheroid formation | HDMEC (Human Dermal Micro-vascular Endothelial Cells), Primary human lung fibroblasts, U87MG (nervous tissue) | [73] |
| PDMS based device | Angiogenesis, OoC | hLFs (human Lung Fibroblasts), HUVECs (Human Umbilical Vein Endothelial Cells) | [74] |
| Two-layered glass-PDMS hybrid system | Spheroid formation, invasion assay, drug screening | U87 (nervous tissue) | [75] |
| 3D-printed system (Vero White Plus FullCure 835 resin) |
Angiogenesis, cell culture, drug screening | bEnd.3 (mouse brain endothelial cell line) | [76] |
| Double-casting of PDMS, with master mold made of PMMA. | Spheroid formation, drug screening | Caco-2 (Colon), NHDF (Normal Human Dermal Fibroblast), HepG2 (liver), A549 (lung) | [77] |
| 3D-hydrogel device | Drug screening, OoC | hCMEC/D3 (endothelial cell), HUVECs (Human Umbilical Vein Endothelial Cells), primary neurons, astrocytes | [78] |
| PDMS based device | OoC, drug screening | C3A (liver), EA.hy926 (endothelial) | [79] |
| PMMA-PDMS hybrid system and bioprinted hydrogel scaffold | OoC, angiogenesis | HUVECs (Human Umbilical Vein Endothelial Cells), neonatal rate cardiomyocytes | [80] |
| PDMS based device | OoC, disease model, drug screening | hiPSCs (human induced Pluripotent Stem Cells), CMs (Cardiomyocytes) differentiated from hiPSCs | [81] |