Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Tomoki Maekawa and Version 2 by Rita Xu.
Periodontitis is one of the most common oral diseases resulting in gingival inflammation and tooth loss. Growing evidence indicates that it results from dysbiosis of the oral microbiome, which interferes with the host immune system, leading to bone destruction. Immune cells activate periodontal ligament cells to express the receptor activator of nuclear factor kappa-B (NF-κB) ligand (RANKL) and promote osteoclast activity. Osteocytes have active roles in periodontitis progression in the bone matrix. Local proteins are involved in bone regeneration through functional immunological plasticity.
- periodontitis
- osteoimmunology
- bone regeneration
- osteoblast
- osteocyte
- osteoclast
1. Introduction
The human oral cavity contains many microbial species constituting commensal bacteria, with interactions between the host and oral microbiota defining oral cavity health. The oral microbiota is a distinct and diversified ecosystem of microbial organisms that each microorganism interacts with, metabolically and physically. These complex interactions culminate in the creation of biofilm communities in which physiochemical gradients establish diverse habitats for microorganisms requiring varying metabolic activities [1]. The microbiota structure in healthy supragingival plaque resembles the “hedgehog-like” network, resulting from the radially spatio-chemical gradient [2]. In the structure depicting the plaque microbiota described by Welch et al. [2], early colonizers, such as Actinomyces spp. and Streptococcus spp., adhere to the tooth surface via interaction of non-specific and specific binding adhesins on their cell surfaces and salivary proteins within the pellicle [3]. Consequently, Corynebacterium spp. attaches to the early colonizers and radially grows exteriorly to form a long, annular structure. Haemophilus, Aggregatibacter, and Neisseriaceae attach to the tip of the annulus owing to their metabolic activity requiring the abundance of both oxygen and nutrients. Metabolic products from oxidative species at the periphery create anoxic circumstances in the core of the biofilm, where anoxic capnophilic species, such as Capnocytophaga and Fusobacterium, prefer to grow [2]. Unlike many infectious diseases, periodontitis results from oral microbiota dysbiosis [4][5][6][4,5,6]. The etiology of periodontal diseases is due to periodontal-pathogenic bacteria. When the sophisticated interactions of the oral microbiota are disturbed by the keystone bacterium, Porphyromonas gingivalis, the polymicrobial dysbiosis transpires [7][8][7,8]. This disturbance in plaque microbiota is a significant etiology of gingival inflammation causing the onset of periodontitis [8][9][10][8,9,10]. Oral microbiota dysbiosis interferes with the host immune system, resulting in inflammatory conditions, in turn leading to cellular interactions of immune and bone cells and subsequent bone destruction [11][12][11,12]. The cellular interactions and molecules related to the communication between immune and bone cells have gained popularity in recent years, providing more insights into periodontitis.
Osteoimmunology focuses on the cellular and molecular mechanisms behind inflammatory bone resorption, which destroys alveolar bone [13]. Recent studies in osteoimmunology focused on understanding the pathogenesis and development of therapies for inflammatory bone diseases, such as rheumatoid arthritis and periodontitis [14].
2. Periodontitis: Osteoimmunology
2.1. RANKL and OPG
Bone is a dynamic tissue resulting from calcium and collagen homeostasis through balanced osteoclast and osteoblast function. Osteoclasts derived from monocytes/macrophages are multinucleated cells that resorb bone matrix. Osteoblasts control osteoclastogenesis via the production of macrophage colony-stimulating factor (M-CSF) and RANKL, an essential cytokine in the activation of osteoclasts [14][15][16][14,24,70]. Alveolar loss results from dominant osteoclast activity. This activity depends on the interaction of three proteins composing the RANK/RANKL/OPG axis [17][18][71,72]. Transmembrane receptor activator of nuclear factor-κB (RANK) receptor is expressed in progenitor and mature osteoclasts with RANK binding to its ligand, RANKL, determining osteoclast activity. RANKL is a cytokine in the TNF family [13] that activates the RANK receptor [19][73]. However, osteoprotegerin (OPG) is a soluble decoy receptor for RANKL, impeding the RANK/RANKL binding and inhibiting osteoclastogenesis [20][74]. The RANKL/OPG ratio increases at periodontitis-affected sites, emphasizing the importance of the equilibrium between the molecules in this axis, especially RANKL and OPG levels [21][22][75,76]. Membrane-bound RANKL is responsible for almost all primary functions; however, soluble RANKL has a minor role in physiological bone homeostasis in mice [23][77]. Depriving soluble RANKL in the mouse model did not influence the level of bone destruction, which emphasizes the role of membrane-bound RANKL in the pathology of osteoporosis and periodontitis [15][23][24][24,77,78]. Accordingly, cells expressing membrane-bound RANKL are either near the bone surface or in contact with it [14]. exFoxp3Th17 cells exhibit the highest mRNA and protein levels of RANKL amongst T cell subsets [15][24]. Moreover, Th17 and exFoxp3Th17 cells produce IL-17 to activate mesenchymal cells, such as osteoblasts and PDL cells, to express RANKL on their membrane and produce proinflammatory cytokines to stimulate osteoclastic differentiation following alveolar bone loss [15][25][24,79]. Therefore, cells expressing membrane-bound RANKL derived near the periodontitis-affected bone surface lead to periodontitis. In addition, Th17 cells and exFoxp3Th17 T cell subsets likely contribute to mesenchymal cell stimulation via IL-17 to express RANKL, followed by stimulation of osteoclast functions. Reduced OPG levels result in an elevated RANKL/OPG ratio, with a human study demonstrating significantly decreased OPG mRNA expression and OPG immunostaining in periodontitis lesions compared with the healthy periodontium [26][80]. In addition, proteases derived from oral bacteria and osteoclasts cleave OPG and stimulate osteoclast function in vitro [27][28][81,82]. This emphasizes the important roles of each protein in controlling osteoclast function in the axis, which is the critical activity affecting periodontitis.2.2. Osteocytes Are Not Just Quiescent Resident Cells
Periodontitis is a multidimensional disease causing the tissue and teeth-supporting bone to disorganize. The pathophysiological process of periodontal disease has primarily focused on bone loss, particularly osteoclastogenesis and osteoblastogenesis. Recent studies show that osteocytes are not quiescent and contribute to physiological and pathological events in periodontitis [29][83]. Osteocytes are derived from an osteoblast cell lineage differentiated from mesenchymal stem cells (MSCs) in the bone marrow and account for most of the bone cells. They have an extended lifetime of up to 25 years compared with a half-life of 150 days for osteoblasts [30][84]. When osteoblasts cease to secrete extracellular matrix (ECM), they either undergo apoptosis or differentiate into osteocytes [31][32][85,86]. Communication between osteocytes and other cells can occur via dendrites reaching the surface of the bone (the lacunocanalicular system) [32][86]. This connection establishes crosstalk between osteoclasts, osteoblasts, bone lining cells, and bone marrow cells with the osteocytes [30][32][33][84,86,87]. Osteoclasts affect bone metabolism via osteocyte connectors. Osteoclast-derived leukemia inhibitory factor reduces sclerostin expression in osteocytes and subsequently promotes osteoblastic bone formation [34][35][88,89].2.3. Osteocytes Induce Osteoclastogenesis via M-CSF (CSF-1) and RANKL
Osteoclasts are multinucleated giant cells originating from monocyte/macrophage cells and are accountable for bone resorption. Characteristics of periodontitis progression include alveolar bone loss resulting from osteoclast differentiation or osteoclastogenesis [36][90]. Osteoclastogenesis occurs via the production of colony-stimulating factor-1, also known as M-CSF, a factor required for osteoclast differentiation [37][38][91,92]. Continuous production of M-CSF in osteocytes enhances osteoclastogenesis [39][93]. Osteocyte-derived M-CSF protects against excessive Nox4-derived ROS generation and retains bone remodeling [40][94]. RANKL is a membrane-associated cytokine required for osteoclastogenesis synthesized by osteoblasts and osteocytes [37][91]. Osteoclastogenesis driven by RANKL is critical for inflammatory bone resorption, and its expression increases in periodontitis [41][42][95,96]. RANKL is synthesized by osteoblasts and interacts with the RANK receptor on the cell membrane of pre-osteoclasts to initiate conversion into active osteoclasts. OPG is the naturally present RANKL decoy receptor and is generated by various osteoblast lineages [43][97]. OPG competes with and reduces RANKL binding to RANK, inhibiting the activation of osteoclasts [44][98]. Osteocytes produce RANKL during bone remodeling in periodontitis (Figure 1). The concept of osteoblasts being the primary cellular source of RANKL has shifted toward osteocytes, which serve as the primary source of RANKL in bone remodeling instead of osteoblasts [45][46][99,100]. Gram-negative bacteria-derived lipopolysaccharide interacts with toll-like receptors on the osteocyte cell surface to stimulate the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) 1/2 signaling pathway resulting in the activation of transcription factors that upregulate IL-6 expression [47][101]. IL-6 then activates Janus kinase through gp130, phosphorylating signaling molecules, and activator of transcription (STAT), leading to the translocation of STAT into the nucleus, enhancing RANKL expression in osteocytes [48][49][102,103]. Apart from IL-6, TNF-α is a typical inflammatory cytokine in periodontitis that directly stimulates osteocytes to produce RANKL and induce osteoclastogenesis or promote sclerostin expression in osteocytes leading to increased osteoclastogenesis [39][93]. Nevertheless, TNF-α does not induce osteocytes to produce M-CSF [50][104].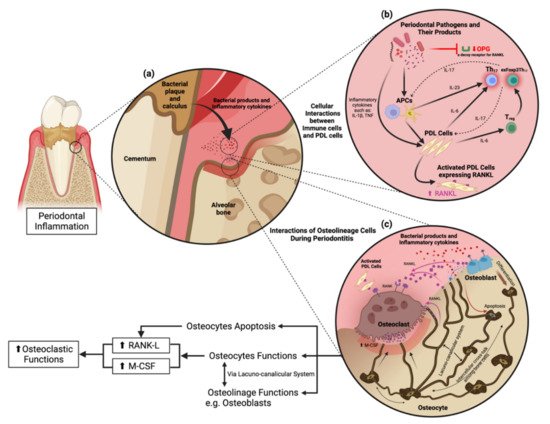
Figure 1. Cellular interactions and osteocyte function in periodontitis. (a) Pathogenic bacteria in the periodontal pocket release their virulence factors to stimulate proinflammatory cytokine production from periodontal stromal cells and immune cells in the periodontium. (b) Cellular interactions in response to periodontitis. Periodontal pathogens and their products activate antigen-presenting cells (APCs) in the periodontium to stimulate periodontal ligament cells (PDL cells) following the activation of Th17 and exFoxp3Th17 cells. (c) Osteocyte functions and their interactions with other cells through the lacunocanalicular system (LCS).