Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Noriaki Emoto.
Endothelin was first discovered more than 30 years ago as a potent vasoconstrictor. In subsequent years, three isoforms, two canonical receptors, and two converting enzymes were identified, and their basic functions were elucidated by numerous preclinical and clinical studies. The endothelin system has been found to be critical in the pathogenesis of several cardiovascular diseases, including hypertension, pulmonary arterial hypertension, heart failure, and coronary artery disease.
- endothelin
- endothelin receptor antagonist
- pulmonary hypertension
- heart failure
- coronary artery disease
- hypertension
1. Biosynthesis of Endothelin
ET-1 belongs to the most abundantly synthesized endothelin peptide family. Mature ET-1 is a 21-amino-acid peptide with two cysteine bridges at the N-terminus and a free hydrophobic C-terminus. The crystal structure of ET-1 was solved recently using X-ray diffraction data collected in 1992 [17,18][1][2]. Endothelins have structures similar to snake venom toxins (safarotoxins), whose envenomation causes strong coronary artery constriction [19,20][3][4]. Endothelin receptor antagonists have been suggested as antivenoms [21][5]. Mature ET-1 peptide is synthetized by many types of cells, mainly vascular endothelial and smooth muscle cells, while macrophages, fibroblasts, podocytes, and brain neurons also express it [2,13][6][7]. Meanwhile, ET-2 peptide is synthetized mainly by intestinal epithelial cells, while it is also transiently expressed in the lung and ovarian follicles [7,22,23][8][9][10]. Finally, the ET-3 peptide is synthetized by melanocytes, intestinal cells, brain neurons, and other cells [2,24,25][6][11][12]. Endothelin peptide synthesis is activated in response to many factors such as hyperglycemia, hypercholesterolemia, aging, estrogen deficiency, hypoxia, shear stress, microRNAs, and angiotensin II [22,23,24,25][9][10][11][12].
Endothelin biosynthesis involves three steps, as illustrated in Figure 1. Endothelins are initially secreted as precursor 212 amino acid polypeptides, named preproETs. A signal peptidase cleaves the 17-amino acid signal to generate proETs, which are subsequently cleaved at the C and N terminals by furin enzymes to generate big ETs [25,26][12][13]. Finally, endothelin-converting enzymes (ECEs) cleave big ETs to produce mature ETs with 21 amino acids [14]. Because big ETs are biologically inactive, this maturation process is their key activity. Interestingly, in mice lacking both ECE-1 and ECE-2, mature endothelin peptide levels were reduced by one-third [27][15]. Other enzymes such as chymases are involved in the maturation of big ETs [28,29][16][17]. The deletion of chymases reduces mature endothelin levels [30[18][19],31], whereas overexpression increases it [32,33][20][21].
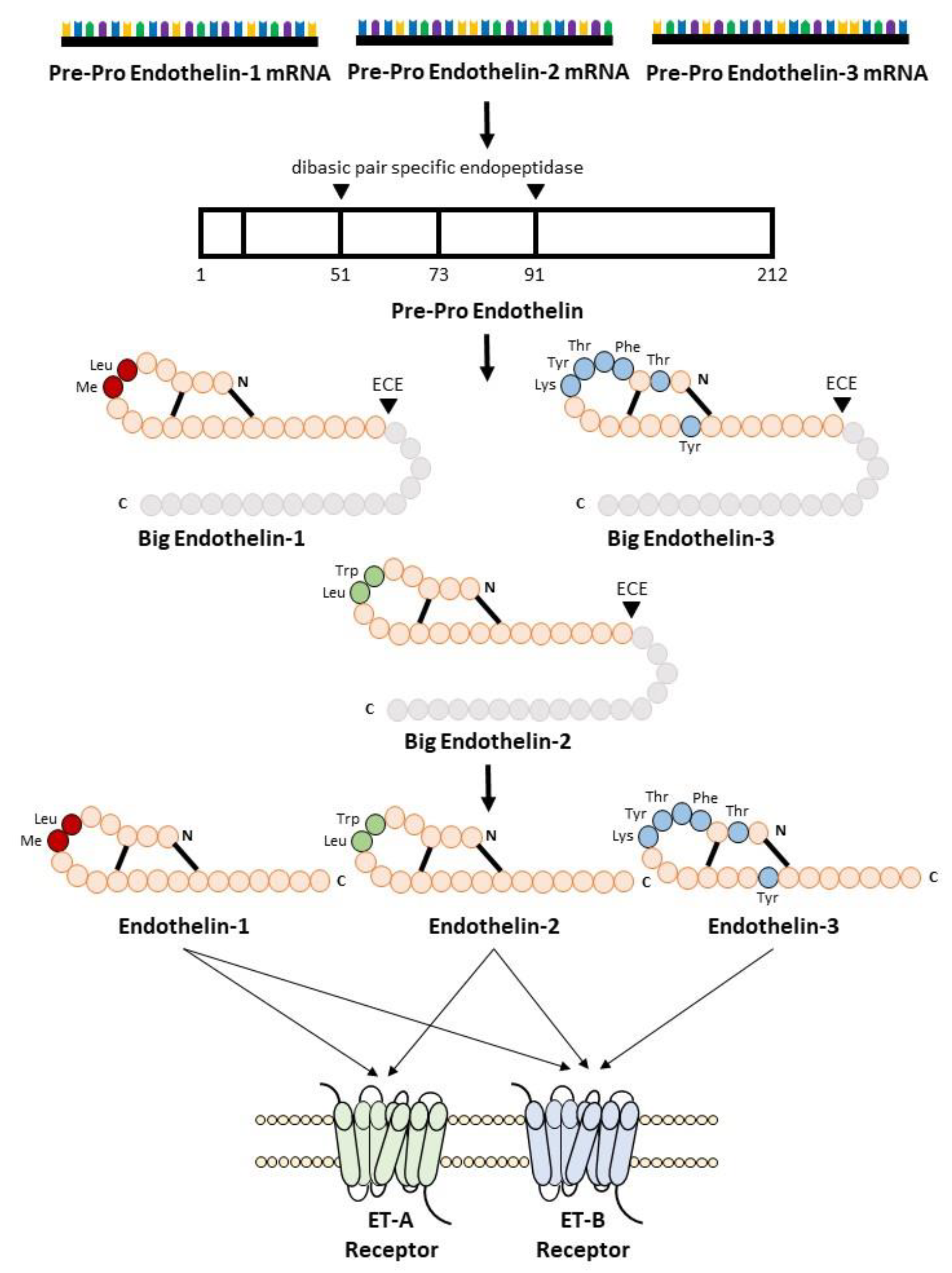
Figure 1.
Biosynthesis of endothelin.
2. Endothelin Receptor
To activate its signaling pathways, the endothelin peptides bind to two subtypes of endothelin receptor, the ETA receptor [9,10][22][23] and the ETB receptor [11[24][25],12], which belong to the seven G-protein-coupled transmembrane-spanning domain receptors (GPCRs). Both ET-1 and ET-2 showed equal potency for the ETA receptor binding, whereas ET-3 showed 100-fold lower affinity for the ETA receptor. In contrast, ET-1, ET-2, and ET-3 showed similar potency to ETB receptors [13,34][7][26]. ETA receptor expression was relatively higher in the vascular smooth muscle, whereas ETB receptor expression was higher in endothelial cells. Thus, ETA and ETB receptors are ubiquitously expressed in all organs that receive the blood supply. The ETA receptor was expressed at the highest level in the lungs and heart, with lower expression in the brain, while the brain and periphery of the lung, such as capillaries, are rich in ETB receptors [35][27].
ETA receptor stimulation induced potent and prolonged vasoconstriction, inflammation, and cell proliferation, whereas ETB receptor stimulation generally showed the opposite effects (see Figure 2) [34,36][26][28]. As such, the ETB receptor can be considered an ETA receptor endogenous antagonist. The ETB receptor also functions in the clearance of ET-1 from circulation (see Figure 2) [37,38,39][29][30][31]. The crystal structure of the ETB receptor and its interaction with ligands have been recently determined [40,41,42,43,44][32][33][34][35][36]. These findings shed light on the interaction between the ETB receptor and its ligand as well as the underlying G-protein mechanism.
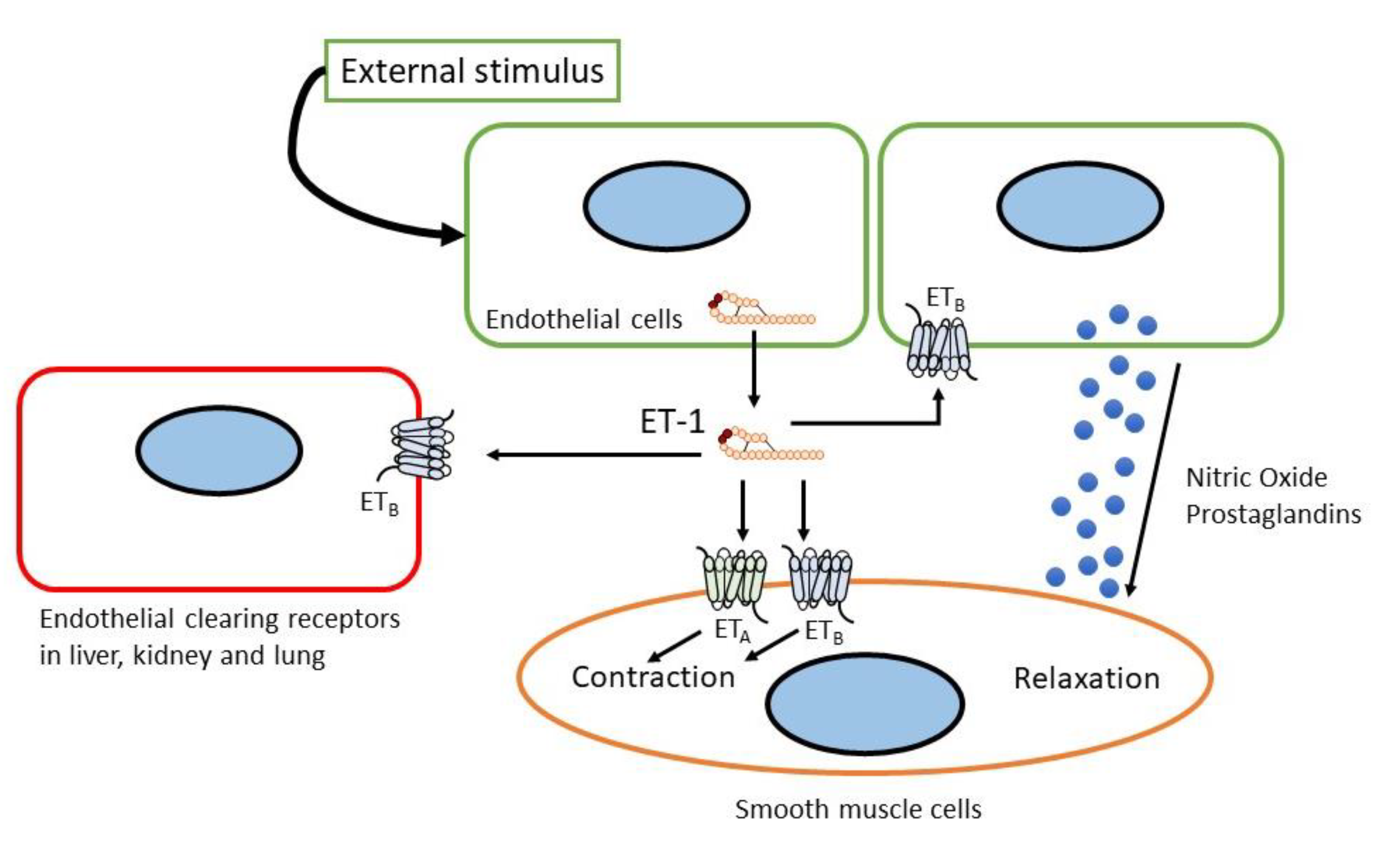
Figure 2.
Sites and mechanism of action of endothelin.
3. Endothelin Agonists and Antagonists
Numerous peptide and non-peptide compounds that act on endothelin receptors with varying degrees of potency and specificity have been discovered. Some of these compounds act as agonists and antagonists. Several compounds can act selectively, while others are non-selective on endothelin receptors [13][7]. Over the last two decades, the development of agonists and antagonists for endothelin receptors, ETA and ETB, has been extensively studied. BQ123 and FR139317 were the first ETA-selective peptide antagonists to be identified. Parallelly, the ETB agonists (BQ3020 and IRL1620) and the first selective antagonist peptide ETB (BQ788) were identified. Within five years of the discovery of ET-1, a bioavailable non-peptide antagonist drug of the endothelin system was developed.
ET-1, ET-2, and ET-3 are agonists of the ETA and ETB receptors. However, because ET-3 has a lower affinity for the ETA receptor, it is more likely to activate the ETB receptor [45][37]. To date, no ETA receptor agonists, either peptides or non-peptides, have been identified. It is generally accepted that the effects of ETA activation in pathophysiological conditions are deleterious; therefore, there is no beneficial evidence for activating the ET-1/ETA pathway [46][38]; however, several ETB receptor agonists have been discovered to date. Sarafotoxin 6c, which has been used in experimental studies in humans, has notably high selectivity for rat ETB receptors, but less so for human ETB receptors [47,48][39][40]. IRL1620 [49][41] and BQ3020 [50][42] are the most widely used selective ETB receptors. IRL1620 is used in experiments involving cerebral blood flow as a neuroprotective agent [51,52,53][43][44][45] and in cancers [54,55,56][46][47][48]. BQ3020 has been used in ETB receptor characterization and labeling studies [57,58,59][49][50][51] and as a selective PET agent in vivo [60][52]. However, there is currently no evidence that agonist agents of endothelin have been initiated in cardiology.
On the contrary, endothelin receptor antagonists (ERAs) have been identified and utilized for several years. ERAs are classified as selective towards one receptor subtype or dual antagonists that block both ETA and ETB receptors. There is no agreement regarding the classification of these antagonists; however, Davenport and Maguire suggested that selective compounds should have more than 100-fold selectivity towards either ETA or ETB receptors, while those that display less selectivity than that are defined as balanced antagonists [61][53]. The clinical evidence of ERA use in cardiovascular diseases will be discussed in a later section.
Bosentan is the first antagonist of both ETA and ETB receptors and was approved by the U.S. Food and Drug Administration in 2001 for pulmonary arterial hypertension (PAH) [62,63][54][55]. The diverse side-effects of bosentan include headache, nasal congestion, flushing, fluid edema, elevated levels of liver enzymes, and anemia, which resemble those of ETA-selective antagonists. Bosentan-related elevation of liver enzymes is dose-dependent and typically asymptomatic [13,64,65,66][7][56][57][58]. Macitentan is a non-selective endothelin receptor antagonist that was approved for clinical use in PAH in 2013. It was designed by modifying the structure of bosentan to improve its efficacy and tolerability, resulting in reduced side effects, such as lower liver toxicity and lower extremity fluid retention, compared to bosentan [61][53]. Procitentan is a potent dual ET receptor antagonist derived from macitentan. Aprocitentan is currently under investigation for treatment-resistant hypertension, which will be discussed in detail later [67,68][59][60].
The most widely used ETA receptor selective antagonist is BQ123 [69][61] which has been used in both in vivo and in vitro studies. Other peptide-based selective ETA receptor antagonists used in experiments were FR139317 [70][62] and TAK-044 [71][63]. Ambrisentan [72][64] and sitaxentan [73][65] have been used in clinical trials to treat PAH patients. Ambrisentan was the second approved antagonist introduced in clinical settings for PAH treatment in 2007. However, in 2010, sixatentan was withdrawn owing to cases of idiosyncratic hepatitis resulting in acute liver failure and death [74][66]. Atrasentan [75][67], another highly selective ETA receptor antagonist, has been successfully used in the treatment of diabetic nephropathy [76][68].
Selective ETB receptor antagonists are less developed compared to other types of endothelin receptor antagonists, attributed to the potential danger of blocking ET-1 clearance and vasodilatation effects [13][7]. In the pre-clinical setting, the most extensively used ETB antagonist is peptide BQ788 [77][69]. The last novel derivative from ERA is the relatively novel agent, sparcentan. Sparcentan is the first orally active antagonist with ETA receptor and angiotensin II type 1 (AT1) receptor inhibitory activities in a single compound. It was developed by merging the elements present in the irbesartan AT1 receptor antagonists with elements in biphenylsulfonamide ETA receptors. Currently, sparcentan has been investigated in several clinical trials related to kidney diseases [46,78][38][70].
4. Genetic Mutations in Endothelin System
Genetic mutations in endothelins, endothelin converting enzymes, and endothelin receptors have been shown to be involved in or risk factors for many diseases. For instance, mutations in endothelin 1 gene are associated with pediatric pulmonary hypertension [79][71], recessive auriculocondylar syndrome (ACS), and dominant isolated question-mark ears (QME) [80][72]. The rs9349379 SNP of the PHACTR1 locus (6p24), which is associated with coronary artery disease (CAD), migraine headache, cervical artery dissection, fibromuscular dysplasia, and systemic arterial hypertension [81][73], is a regulator of endothelin-1 expression [82][74].
Mutation in the ETA receptor peptide-binding site alters its subtype selectivity, which affects its interaction with ligands [83][75]. Mutations in the ETA receptor cause mandibulofacial dysostosis with alopecia [84][76]. The genetic variant of EDNRA, rs6841581, is significantly associated with an increased risk of intracranial aneurysm in East Asian populations [85,86,87][77][78][79]. The ETA receptor (ENDRA-231 A/G) gene polymorphism is associated with migraine [88,89][80][81].
Mutations in endothelin-3 commonly affect the enteric nervous system and the melanocytes. As endothelin-3 exerts its function by interacting with the ETB receptor, a similar phenomenon occurred in ETB receptor mutations. Several mutations in endothelin-3 are associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome) [90,91,92][82][83][84]. Mutations in the ETB receptor are also associated with Hirschsprung and Waardenburg syndromes [93,94,95,96][85][86][87][88]. Hypermethylation and downregulation of the ETB receptor expression are associated with reduced patient survival and poor prognosis in several types of malignancies [97,98,99,100][89][90][91][92].
Not limited to the peptides or receptors only, mutations in the converting enzymes have also been linked to pathological conditions. The R742C mutation in the ECE-1 gene results in a patient with skip lesion Hirschsprung disease, cardiac defects, and autonomic dysfunction [101][93]. Another variation in ECE-1 is linked to essential hypertension [102][94].
5. Phenotype of Genetic Endothelin Modification in Mice
A whole-body ET-1 knockout mouse was developed by deleting exon 2 of the ET-1 gene [103][95]. Homozygous deletion (ET-1−/−) is lethal in neonates. Caesarian delivered mice on day 18.5, postcoital, all with major craniofacial and cardiac anomalies [103,104][95][96]. ET-1−/− mice also have lower neonatal weight, poor thyroid and thymus development, and lesser cardiac sympathetic innervation [105,106][97][98]. Heterozygous deletion of ET-1 (ET-1+/−) resulted in different phenotypes in which the mice appeared normal, fertile, and with reduced ET-1 concentration in the lung and plasma. However, mice exhibit elevated blood pressure [103][95]. In the overexpression mouse model (ET-1+), the mice exhibited normal ET-1 in the blood, but increased ET-1 expression in the brain, lungs, and kidneys [107][99]. These mice exhibited chronic inflammation in the lungs [107][99]. Kidney phenotypes were more severe, exhibiting increased renal cyst formation, renal interstitial fibrosis, glomerulosclerosis, and age-dependent salt-sensitive hypertension [107,108,109,110][99][100][101][102].
Global ET-2 deletion in mice resulted in severe growth retardation, juvenile lethality, internal starvation, hypothermia, and abnormal lung histology. These findings revealed that ET-2 is important for postnatal growth and survival of mice by regulating energy homeostasis and maintaining lung function [111][103]. Global ET-2 overexpression in Sprague-Dawley rats, called TGR(hET-2)37, results in male rats having significantly lower body weight accompanied by kidney interstitial and glomerular sclerosis. Female rats exhibit glomerulosclerosis [112,113][104][105].
ET-3 heterozygous mice (ET-3+/−) were phenotypically normal. However, global homozygous knockout mice (ET-3−/−) died early postnatally, with an average age of 21 days after birth. The mice also presented with aganglionic megacolon and coat color spotting. This result showed that ET-3 is required for the proper development of enteric neurons derived from the vagal neural crest and epidermal melanocytes derived from the trunk neural crest [114][106]. Piebaldism (absence of melanocytes in the skin) or lethal spotted (ls) phenotypes arose spontaneously in mouse colonies. These ls/ls mice also presented with megacolon. The ET-3 transgene under the control of human dopamine-β-hydroxylase (DβH) introduced into ls/ls mice reduced piebaldism and megacolon in these mice. This evidence shows that the ls/ls mouse phenotype is a result of ET-3 deficiency [115][107].
ETA−/− mice die shortly after birth due to severe craniofacial deformities and neural crest-derived structural abnormalities [116,117][108][109]. ETB+/− mice appeared normal and were able to produce offspring. However, ETB−/− mice were born healthy but became sick and died within 4 weeks, and showed similar abnormalities as ET-3−/− mice, including megacolon and coat color changes [118][110]. ECE-1 deletion resulted in mortality between embryonic day 12.5 (E12.5) and 30 min after birth. ECE-1−/− mice showed cardiac and craniofacial anomalies identical to those in ET-1 and ETA receptor-deficient mice [119][111]. On the other hand, ECE-2−/− mice survive, appear healthy, fertile, and have the same lifespan as wild-type littermates. The simultaneous deletion of ECE-1 and ECE-2 with ECE-1−/−/ECE-2−/− miceshowed broader and more severe cardiac abnormalities than ECE-1−/− mice [27][15].
References
- McPherson, A.; Larson, S.B. The X-ray crystal structure of human endothelin 1, a polypeptide hormone regulator of blood pressure. Acta Crystallogr. Sect. F 2019, 75, 47–53.
- Stanfield, R.L. Never too late for endothelin. Acta Crystallogr. Sect. F 2019, 75, 45–46.
- Kloog, Y.; Ambar, I.; Sokolovsky, M.; Kochva, E.; Wollberg, Z.; Bdolah, A. Sarafotoxin, a Novel Vasoconstrictor Peptide: Phosphoinositide Hydrolysis in Rat Heart and Brain. Science 1988, 242, 268–270.
- Takasaki, C.; Yanagisawa, M.; Kimura, S.; Goto, K.; Masaki, T. Similarity of endothelin to snake venom toxin. Nature 1988, 335, 303.
- Abd-Elsalam, M.A. Bosentan, a selective and more potent antagonist for Atractaspis envenomation than the specific antivenom. Toxicon 2011, 57, 861–870.
- Barton, M.; Yanagisawa, M. Endothelin: 30 years from discovery to therapy. Hypertension 2019, 74, 1232–1265.
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418.
- Saida, K.; Hashimoto, M.; Mitsui, Y.; Ishida, N.; Uchide, T. The Prepro Vasoactive Intestinal Contractor (VIC)/Endothelin-2 Gene (EDN2): Structure, Evolution, Production, and Embryonic Expression. Genomics 2000, 64, 51–61.
- Barton, M. Aging and endothelin: Determinants of disease. Life Sci. 2014, 118, 97–109.
- Stow, L.R.; Jacobs, M.E.; Wingo, C.S.; Cain, B.D. Endothelin-1 gene regulation. FASEB J. 2011, 25, 16–28.
- von Brandenstein, M.; Richter, C.; Fries, J.W.U. MicroRNAs: Small but amazing, and their association with endothelin. Life Sci. 2012, 91, 475–489.
- Houde, M.; Desbiens, L.; D’Orléans-Juste, P. Chapter Five—Endothelin-1: Biosynthesis, Signaling and Vasoreactivity. Adv. Pharmacol. 2016, 77, 143–175.
- Denault, J.-B.; Claing, A.; D’Orléans-Juste, P.; Sawamura, T.; Kido, T.; Masaki, T.; Leduc, R. Processing of proendothelin-1 by human furin convertase. FEBS Lett. 1995, 362, 276–280.
- Xu, D.; Emoto, N.; Giaid, A.; Slaughter, C.; Kaw, S.; deWit, D.; Yanagisawa, M. ECE-1: A membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell 1994, 78, 473–485.
- Yanagisawa, H.; Hammer, R.E.; Richardson, J.A.; Emoto, N.; Williams, S.C.; Takeda, S.; Clouthier, D.E.; Yanagisawa, M. Disruption of ECE-1 and ECE-2 reveals a role for endothelin-converting enzyme-2 in murine cardiac development. J. Clin. Investig. 2000, 105, 1373–1382.
- Wypij, D.M.; Nichols, J.S.; Novak, P.J.; Lowell Stacy, D.; Berman, J.; Wiseman, J.S. Role of mast cell chymase in the extracellular processing of big-endothelin-1 to endothelin-1 in the perfused rat lung. Biochem. Pharmacol. 1992, 43, 845–853.
- Maguire, J.; Davenport, A.P. Alternative Pathway to Endothelin-Converting Enzyme for the Synthesis of Endothelin in Human Blood Vessels. J. Cardiovasc. Pharmacol. 2004, 44, S27.
- Houde, M.; Desbiens, L.; Schwertani, A.; Pejler, G.; Iglarz, M.; D’Orléans-Juste, P. Endothelin receptor antagonist macitentan or deletion of mouse mast cell protease 4 delays lesion development in atherosclerotic mice. Life Sci. 2016, 159, 71–75.
- Houde, M.; Jamain, M.-D.; Labonté, J.; Desbiens, L.; Pejler, G.; Gurish, M.; Takai, S.; D’Orléans-Juste, P. Pivotal Role of Mouse Mast Cell Protease 4 in the Conversion and Pressor Properties of Big-Endothelin-1. J. Pharmacol. Exp. Ther. 2013, 346, 31–37.
- Guo, C.; Ju, H.; Leung, D.; Massaeli, H.; Shi, M.; Rabinovitch, M. A novel vascular smooth muscle chymase is upregulated in hypertensive rats. J. Clin. Investig. 2001, 107, 703–715.
- Ju, H.; Gros, R.; You, X.; Tsang, S.; Husain, M.; Rabinovitch, M. Conditional and targeted overexpression of vascular chymase causes hypertension in transgenic mice. Proc. Natl. Acad. Sci. USA 2001, 98, 7469–7474.
- Arai, H.; Hori, S.; Aramori, I.; Ohkubo, H.; Nakanishi, S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature 1990, 348, 730–732.
- Sakurai, T.; Yanagisawa, M.; Takuwat, Y.; Miyazakit, H.; Kimura, S.; Goto, K.; Masaki, T. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 1990, 348, 732–735.
- Sakamoto, A.; Yanagisawa, M.; Sakurai, T.; Takuwa, Y.; Yanagisawa, H.; Masaki, T. Cloning and functional expression of human cDNA for the ETB endothelin receptor. Biochem. Biophys. Res. Commun. 1991, 178, 656–663.
- Saito, Y.; Mizuno, T.; Itakura, M.; Suzuki, Y.; Ito, T.; Hagiwara, H.; Hirose, S. Primary structure of bovine endothelin ETB receptor and identification of signal peptidase and metal proteinase cleavage sites. J. Biol. Chem. 1991, 266, 23433–23437.
- Dhaun, N.; Webb, D.J. Endothelins in cardiovascular biology and therapeutics. Nat. Rev. Cardiol. 2019, 16, 491–502.
- Regard, J.B.; Sato, I.T.; Coughlin, S.R. Anatomical Profiling of G Protein-Coupled Receptor Expression. Cell 2008, 135, 561–571.
- Schneider, M.P.; Boesen, E.I.; Pollock, D.M. Contrasting Actions of Endothelin ETA and ETB Receptors in Cardiovascular Disease. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 731–759.
- Dupuis, J.; Goresky, C.A.; Fournier, A. Pulmonary clearance of circulating endothelin-1 in dogs in vivo: Exclusive role of ETBreceptors. J. Appl. Physiol. 1996, 81, 1510–1515.
- Dupuis, J.; Stewart, D.J.; Cernacek, P.; Gosselin, G. Human Pulmonary Circulation Is an Important Site for Both Clearance and Production of Endothelin-1. Circulation 1996, 94, 1578–1584.
- Fukuroda, T.; Fujikawa, T.; Ozaki, S.; Ishikawa, K.; Yano, M.; Nishikibe, M. Clearance of Circulating Endothelin-1 by ETB Receptors in Rats. Biochem. Biophys. Res. Commun. 1994, 199, 1461–1465.
- Shihoya, W.; Nishizawa, T.; Okuta, A.; Tani, K.; Dohmae, N.; Fujiyoshi, Y.; Nureki, O.; Doi, T. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 2016, 537, 363–368.
- Shihoya, W.; Izume, T.; Inoue, A.; Yamashita, K.; Kadji, F.M.N.; Hirata, K.; Aoki, J.; Nishizawa, T.; Nureki, O. Crystal structures of human ETB receptor provide mechanistic insight into receptor activation and partial activation. Nat. Commun. 2018, 9, 4711.
- Shihoya, W.; Nishizawa, T.; Yamashita, K.; Inoue, A.; Hirata, K.; Kadji, F.M.N.; Okuta, A.; Tani, K.; Aoki, J.; Fujiyoshi, Y.; et al. X-ray structures of endothelin ETB receptor bound to clinical antagonist bosentan and its analog. Nat. Struct. Mol. Biol. 2017, 24, 758–764.
- Nagiri, C.; Shihoya, W.; Inoue, A.; Kadji, F.M.N.; Aoki, J.; Nureki, O. Crystal structure of human endothelin ETB receptor in complex with peptide inverse agonist IRL2500. Commun. Biol. 2019, 2, 236.
- Izume, T.; Miyauchi, H.; Shihoya, W.; Nureki, O. Crystal structure of human endothelin ETB receptor in complex with sarafotoxin S6b. Biochem. Biophys. Res. Commun. 2020, 528, 383–388.
- Haynes, W.G.; Strachan, F.E.; Webb, D.J. Endothelin ETA and ETB Receptors Cause Vasoconstriction of Human Resistance and Capacitance Vessels In Vivo. Circulation 1995, 92, 357–363.
- Davenport, A.P.; Kuc, R.E.; Southan, C.; Maguire, J.J. New drugs and emerging therapeutic targets in the endothelin signaling pathway and prospects for personalized precision medicine. Physiol. Res. 2018, 67, S37–S54.
- Russell, F.D.; Davenport, A.P. Characterization of the binding of endothelin ETB selective ligands in human and rat heart. Br. J. Pharmacol. 1996, 119, 631–636.
- Williams, D.L.; Jones, K.L.; Pettibone, D.J.; Lis, E.V.; Clineschmidt, B.V. Sarafotoxin S6c: An agonist which distinguishes between endothelin receptor subtypes. Biochem. Biophys. Res. Commun. 1991, 175, 556–561.
- Takai, M.; Umemura, I.; Yamasaki, K.; Watakabe, T.; Fujitani, Y.; Oda, K.; Urade, Y.; Inui, T.; Yamamura, T.; Okada, T. A potent and specific agonist, Suc--endothelin-1(8-21), IRL 1620, for the ETB receptor. Biochem. Biophys. Res. Commun. 1992, 184, 953–959.
- Ihara, M.; Saeki, T.; Fukuroda, T.; Kimura, S.; Ozaki, S.; Patel, A.C.; Yano, M. A novel radioligand BQ-3020 selective for endothelin (ETB) receptors. Life Sci. 1992, 51, PL47–PL52.
- Leonard, M.G.; Briyal, S.; Gulati, A. Endothelin B receptor agonist, IRL-1620, reduces neurological damage following permanent middle cerebral artery occlusion in rats. Brain Res. 2011, 1420, 48–58.
- Cifuentes, E.G.; Hornick, M.G.; Havalad, S.; Donovan, R.L.; Gulati, A. Neuroprotective Effect of IRL-1620, an Endothelin B Receptor Agonist, on a Pediatric Rat Model of Middle Cerebral Artery Occlusion. Front. Pediatr. 2018, 6, 310.
- Briyal, S.; Ranjan, A.K.; Hornick, M.G.; Puppala, A.K.; Luu, T.; Gulati, A. Anti-apoptotic activity of ETB receptor agonist, IRL-1620, protects neural cells in rats with cerebral ischemia. Sci. Rep. 2019, 9, 10439.
- Cemazar, M.; Wilson, I.; Prise, V.E.; Bell, K.M.; Hill, S.A.; Tozer, G.M. The endothelin B (ETB) receptor agonist IRL 1620 is highly vasoconstrictive in two syngeneic rat tumour lines: Potential for selective tumour blood flow modification. Br. J. Cancer 2005, 93, 98–106.
- Rajeshkumar, N.V.; Rai, A.; Gulati, A. Endothelin B receptor agonist, IRL 1620, enhances the anti-tumor efficacy of paclitaxel in breast tumor rats. Breast Cancer Res. Treat. 2005, 94, 237–247.
- Gulati, A.; Sunila, E.; Kuttan, G. IRL-1620, an Endothelin-B Receptor Agonist, Enhanced Radiation Induced Reduction in Tumor Volume in Dalton’s Lymphoma Ascites Tumor Model. Arzneimittelforschung 2012, 62, 14–17.
- Perreault, T.; Baribeau, J. Characterization of endothelin receptors in newborn piglet lung. Am. J. Physiol. Cell. Mol. Physiol. 1995, 268, L607–L614.
- Hirata, Y.; Emori, T.; Eguchi, S.; Kanno, K.; Imai, T.; Ohta, K.; Marumo, F. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J. Clin. Investig. 1993, 91, 1367–1373.
- Molenaar, P.; O’Reilly, G.; Sharkey, A.; Kuc, R.E.; Harding, D.P.; Plumpton, C.; Gresham, G.A.; Davenport, A.P. Characterization and localization of endothelin receptor subtypes in the human atrioventricular conducting system and myocardium. Circ. Res. 1993, 72, 526–538.
- Johnström, P.; Rudd, J.H.F.; Richards, H.K.; Fryer, T.D.; Clark, J.C.; Weissberg, P.L.; Pickard, J.D.; Davenport, A.P. Imaging Endothelin ETB Receptors Using -BQ3020: In Vitro Characterization and Positron Emission Tomography (MicroPET). Exp. Biol. Med. 2006, 231, 736–740.
- Maguire, J.J.; Davenport, A.P. Endothelin Receptors and Their Antagonists. Semin. Nephrol. 2015, 35, 125–136.
- Palmer, M.J. Endothelin Receptor Antagonists: Status and Learning 20 Years On. In Progress in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2009; ISBN 0079-6468.
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2002, 346, 896–903.
- Abman, S.H. Role of Endothelin Receptor Antagonists in the Treatment of Pulmonary Arterial Hypertension. Annu. Rev. Med. 2009, 60, 13–23.
- Maguire, J.J.; Davenport, A.P. —New agonists, antagonists, inhibitors and emerging research frontiers: IUPHAR Review 12. Br. J. Pharmacol. 2014, 171, 5555–5572.
- Serasli, E.; Michaelidis, V.; Kosmas, A.; Antoniadou, M.; Tsara, V. Review on bosentan, a dual endothelin receptor antagonist for the treatment of pulmonary arterial hypertension. Recent Pat. Cardiovasc. Drug Discov. 2010, 5, 184–195.
- Angeli, F.; Verdecchia, P.; Reboldi, G. Aprocitentan, A Dual Endothelin Receptor Antagonist Under Development for the Treatment of Resistant Hypertension. Cardiol. Ther. 2021, 10, 397–406.
- Verweij, P.; Danaietash, P.; Flamion, B.; Ménard, J.; Bellet, M. Randomized dose-response study of the new dual endothelin receptor antagonist aprocitentan in hypertension. Hypertension 2020, 75, 956–965.
- Ihara, M.; Fukuroda, T.; Saeki, T.; Nishikibe, M.; Kojiri, K.; Suda, H.; Yano, M. An endothelin receptor (ETA) antagonist isolated from Streptomyces misakiensis. Biochem. Biophys. Res. Commun. 1991, 178, 132–137.
- Aramori, I.; Nirei, H.; Shoubo, M.; Sogabe, K.; Nakamura, K.; Kojo, H.; Notsu, Y.; Ono, T.; Nakanishi, S. Subtype selectivity of a novel endothelin antagonist, FR139317, for the two endothelin receptors in transfected Chinese hamster ovary cells. Mol. Pharmacol. 1993, 43, 127–131.
- Masuda, Y.; Sugo, T.; Kikuchi, T.; Kawata, A.; Satoh, M.; Fujisawa, Y.; Itoh, Y.; Wakimasu, M.; Ohtaki, T. Receptor binding and antagonist properties of a novel endothelin receptor antagonist, TAK-044 -L-alanyl-L-alpha-aspartyl-D-2-(2-thienyl) glycyl-L-leucyl-D-tryptophyl]disodium salt], in human end. J. Pharmacol. Exp. Ther. 1996, 279, 675–685.
- Vatter, H.; Zimmermann, M.; Weyrauch, E.; Lange, B.N.; Setzer, M.; Raabe, A.; Seifert, V. Cerebrovascular Characterization of the Novel Nonpeptide Endothelin-A Receptor Antagonist LU 208075. Clin. Neuropharmacol. 2003, 26, 73–83.
- Wu, C.; Chan, M.F.; Stavros, F.; Raju, B.; Okun, I.; Mong, S.; Keller, K.M.; Brock, T.; Kogan, T.P.; Dixon, R.A.F. Discovery of TBC11251, a Potent, Long Acting, Orally Active Endothelin Receptor-A Selective Antagonist. J. Med. Chem. 1997, 40, 1690–1697.
- Don, G.W.; Joseph, F.; Celermajer, D.S.; Corte, T.J. Ironic case of hepatic dysfunction following the global withdrawal of sitaxentan. Intern. Med. J. 2012, 42, 1351–1354.
- Jarvis, M.F.; Wessale, J.L.; Zhu, C.Z.; Lynch, J.J.; Dayton, B.D.; Calzadilla, S.V.; Padley, R.J.; Opgenorth, T.J.; Kowaluk, E.A. ABT-627, an endothelin ETA receptor-selective antagonist, attenuates tactile allodynia in a diabetic rat model of neuropathic pain. Eur. J. Pharmacol. 2000, 388, 29–35.
- Heerspink, H.J.L.; Parving, H.-H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.-F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947.
- Ishikawa, K.; Ihara, M.; Noguchi, K.; Mase, T.; Mino, N.; Saeki, T.; Fukuroda, T.; Fukami, T.; Ozaki, S.; Nagase, T. Biochemical and pharmacological profile of a potent and selective endothelin B-receptor antagonist, BQ-788. Proc. Natl. Acad. Sci. USA 1994, 91, 4892–4896.
- Komers, R.; Diva, U.; Inrig, J.K.; Loewen, A.; Trachtman, H.; Rote, W.E. Study Design of the Phase 3 Sparsentan Versus Irbesartan (DUPLEX) Study in Patients with Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2020, 5, 494–502.
- Kumar, A.; Choudhury, M.; Batra, S.D.; Sikri, K.; Gupta, A. In vivo assessment of a single adenine mutation in 5′UTR of Endothelin-1 gene in paediatric cases with severe pulmonary hypertension: An observational study. BMC Res. Notes 2021, 14, 194.
- Gordon, C.T.; Petit, F.; Kroisel, P.M.; Jakobsen, L.; Zechi-Ceide, R.M.; Oufadem, M.; Bole-Feysot, C.; Pruvost, S.; Masson, C.; Tores, F.; et al. Mutations in Endothelin 1 Cause Recessive Auriculocondylar Syndrome and Dominant Isolated Question-Mark Ears. Am. J. Hum. Genet. 2013, 93, 1118–1125.
- Kiando, S.R.; Tucker, N.R.; Castro-Vega, L.-J.; Katz, A.; D’Escamard, V.; Tréard, C.; Fraher, D.; Albuisson, J.; Kadian-Dodov, D.; Ye, Z.; et al. PHACTR1 Is a Genetic Susceptibility Locus for Fibromuscular Dysplasia Supporting Its Complex Genetic Pattern of Inheritance. PLoS Genet. 2016, 12, e1006367.
- Gupta, R.M.; Hadaya, J.; Trehan, A.; Zekavat, S.M.; Roselli, C.; Klarin, D.; Emdin, C.A.; Hilvering, C.R.E.; Bianchi, V.; Mueller, C.; et al. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell 2017, 170, 522–533.e15.
- Krystek, S.R.; Patel, P.S.; Rose, P.M.; Fisher, S.M.; Kienzle, B.K.; Lach, D.A.; Liu, E.C.; Lynch, J.S.; Novotny, J.; Webb, M.L. Mutation of peptide binding site in transmembrane region of a G protein-coupled receptor accounts for endothelin receptor subtype selectivity. J. Biol. Chem. 1994, 269, 12383–12386.
- Gordon, C.T.; Weaver, K.N.; Zechi-Ceide, R.M.; Madsen, E.C.; Tavares, A.L.P.; Oufadem, M.; Kurihara, Y.; Adameyko, I.; Picard, A.; Breton, S.; et al. Mutations in the Endothelin Receptor Type A Cause Mandibulofacial Dysostosis with Alopecia. Am. J. Hum. Genet. 2015, 96, 519–531.
- Low, S.-K.; Takahashi, A.; Cha, P.-C.; Zembutsu, H.; Kamatani, N.; Kubo, M.; Nakamura, Y. Genome-wide association study for intracranial aneurysm in the Japanese population identifies three candidate susceptible loci and a functional genetic variant at EDNRA. Hum. Mol. Genet. 2012, 21, 2102–2110.
- Yasuno, K.; Bakircioglu, M.; Low, S.-K.; Bilguvar, K.; Gaal, E.; Ruigrok, Y.M.; Niemela, M.; Hata, A.; Bijlenga, P.; Kasuya, H.; et al. Common variant near the endothelin receptor type A (EDNRA) gene is associated with intracranial aneurysm risk. Proc. Natl. Acad. Sci. USA 2011, 108, 19707–19712.
- Hong, E.P.; Kim, B.J.; Jeon, J.P.; Yang, J.S.; Choi, H.J.; Kang, S.H.; Cho, Y.J. Association of Endothelin Receptor Type A with Intracranial Aneurysm in 20,609 East Asians: An Updated Meta-Analysis. World Neurosurg. 2019, 130, e804–e814.
- Tzourio, C.; El Amrani, M.; Poirier, O.; Nicaud, V.; Bousser, M.-G.; Alperovitch, A. Association between migraine and endothelin type A receptor (ETA -231 A/G) gene polymorphism. Neurology 2001, 56, 1273–1277.
- Miao, J.; Wang, F.; Fang, Y. Association of 231G>A polymorphism of endothelin type A receptor gene with migraine: A meta-analysis. J. Neurol. Sci. 2012, 323, 232–235.
- Hofstra, R.M.W.; Osinga, J.; Tan-Sindhunata, G.; Wu, Y.; Kamsteeg, E.-J.; Stulp, R.P.; van Ravenswaaij-Arts, C.; Majoor-Krakauer, D.; Angrist, M.; Chakravarti, A.; et al. A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome). Nat. Genet. 1996, 12, 445–447.
- Edery, P.; Attie, T.; Amiel, J.; Pelet, A.; Eng, C.; Hofstra, R.M.W.; Martelli, H.; Bidaud, C.; Munnich, A.; Lyonnet, S. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat. Genet. 1996, 12, 442–444.
- Pingault, V.; Bondurand, N.; Lemort, N.; Sancandi, M.; Ceccherini, I.; Hugot, J.-P.; Jouk, P.-S.; Goossens, M. A heterozygous endothelin 3 mutation in Waardenburg-Hirschsprung disease: Is there a dosage effect ofEDN3/EDNRB gene mutations on neurocristopathy phenotypes? J. Med. Genet. 2001, 38, 205–209.
- Auricchio, A.; Casari, G.; Staiano, A.; Ballabio, A. Endothelin-B Receptor Mutations in Patients with Isolated Hirschsprung Disease from a Non-Inbred Population. Hum. Mol. Genet. 1996, 5, 351–354.
- Fuchs, S.; Amiel, J.; Claudel, S.; Lyonnet, S.; Corvol, P.; Pinet, F. Functional Characterization of Three Mutations of the Endothelin B Receptor Gene in Patients with Hirschsprung’s Disease: Evidence for Selective Loss of Gi Coupling. Mol. Med. 2001, 7, 115–124.
- Varga, L.; Danis, D.; Drsata, J.; Masindova, I.; Skopkova, M.; Slobodova, Z.; Chrobok, V.; Profant, M.; Gasperikova, D. Novel variants in EDNRB gene in Waardenburg syndrome type II and SOX10 gene in PCWH syndrome. Int. J. Pediatr. Otorhinolaryngol. 2021, 140, 110499.
- Puffenberger, E.G.; Hosoda, K.; Washington, S.S.; Nakao, K.; deWit, D.; Yanagisawa, M.; Chakravarti, A. A missense mutation of the endothelin-B receptor gene in multigenic hirschsprung’s disease. Cell 1994, 79, 1257–1266.
- Wei, F.; Ge, Y.; Li, W.; Wang, X.; Chen, B. Role of endothelin receptor type B (EDNRB) in lung adenocarcinoma. Thorac. Cancer 2020, 11, 1885–1890.
- Smith, S.L.; Damato, B.E.; Scholes, A.G.M.; Nunn, J.; Field, J.K.; Heighway, J. Decreased endothelin receptor B expression in large primary uveal melanomas is associated with early clinical metastasis and short survival. Br. J. Cancer 2002, 87, 1308–1313.
- Knight, L.; Gibson, N.; Bujac, S.; Ellison, G.; Growcott, J.; Brooks, N.; Hughes, A.; Xinarianos, G.; Liloglou, T.; Field, J. Hypermethylation of endothelin receptor type B (EDNRB) is a frequent event in non-small cell lung cancer. Cancer Res. 2007, 67, 1135.
- Tanaka, T.; Sho, M.; Takayama, T.; Wakatsuki, K.; Matsumoto, S.; Migita, K.; Ito, M.; Hamada, K.; Nakajima, Y. Endothelin B receptor expression correlates with tumour angiogenesis and prognosis in oesophageal squamous cell carcinoma. Br. J. Cancer 2014, 110, 1027–1033.
- Hofstra, R.M.W.; Valdenaire, O.; Arch, E.; Osinga, J.; Kroes, H.; Löffler, B.-M.; Hamosh, A.; Meijers, C.; Buys, C.H.C.M. A Loss-of-Function Mutation in the Endothelin-Converting Enzyme 1 (ECE-1) Associated with Hirschsprung Disease, Cardiac Defects, and Autonomic Dysfunction. Am. J. Hum. Genet. 1999, 64, 304–307.
- Funke-Kaiser, H.; Reichenberger, F.; Köpke, K.; Herrmann, S.-M.; Pfeifer, J.; Orzechowski, H.-D.; Zidek, W.; Paul, M.; Brand, E. Differential binding of transcription factor E2F-2 to the endothelin-converting enzyme-1b promoter affects blood pressure regulation. Hum. Mol. Genet. 2003, 12, 423–433.
- Kurihara, Y.; Kurihara, H.; Suzuki, H.; Kodama, T.; Maemura, K.; Nagai, R.; Oda, H.; Kuwaki, T.; Cao, W.-H.; Kamada, N.; et al. Elevated blood pressure and craniofaclal abnormalities in mice deficient in endothelin-1. Nature 1994, 368, 703–710.
- Kurihara, Y.; Kurihara, H.; Oda, H.; Maemura, K.; Nagai, R.; Ishikawa, T.; Yazaki, Y. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J. Clin. Investig. 1995, 96, 293–300.
- Kurihara, Y.; Kurihara, H.; Maemura, K.; Kuwaki, T.; Kumada, M.; Yazaki, Y. Impaired development of the thyroid and thymus in endothelin-1 knockout mice. J. Cardiovasc. Pharmacol. 1995, 26 (Suppl. 3), S13-6.
- Ieda, M.; Fukuda, K.; Hisaka, Y.; Kimura, K.; Kawaguchi, H.; Fujita, J.; Shimoda, K.; Takeshita, E.; Okano, H.; Kurihara, Y.; et al. Endothelin-1 regulates cardiac sympathetic innervation in the rodent heart by controlling nerve growth factor expression. J. Clin. Investig. 2004, 113, 876–884.
- Hocher, B.; Thöne-Reineke, C.; Rohmeiss, P.; Schmager, F.; Slowinski, T.; Burst, V.; Siegmund, F.; Quertermous, T.; Bauer, C.; Neumayer, H.H.; et al. Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J. Clin. Investig. 1997, 99, 1380–1389.
- Hocher, B.; Rohmeiss, P.; Thöne-Reineke, C.; Schwarz, A.; Burst, V.; van der Woude, F.; Bauer, C. Theuring Apoptosis in Kidneys of Endothelin-1 Transgenic Mice. J. Cardiovasc. Pharmacol. 1998, 31, S554–S556.
- Kalk, P.; Thöne-Reineke, C.; Schwarz, A.; Godes, M.; Bauer, C.; Pfab, T.; Hocher, B. Renal phenotype of Et-1 transgenic mice is modulated by androgens. Eur. J. Med. Res. 2009, 14, 55.
- Shindo, T.; Kurihara, H.; Maemura, K.; Kurihara, Y.; Ueda, O.; Suzuki, H.; Kuwaki, T.; Ju, K.-H.; Wang, Y.; Ebihara, A.; et al. Renal damage and salt-dependent hypertension in aged transgenic mice overexpressing endothelin-1. J. Mol. Med. 2002, 80, 105–116.
- Chang, I.; Bramall, A.N.; Baynash, A.G.; Rattner, A.; Rakheja, D.; Post, M.; Joza, S.; McKerlie, C.; Stewart, D.J.; McInnes, R.R.; et al. Endothelin-2 deficiency causes growth retardation, hypothermia, and emphysema in mice. J. Clin. Investig. 2013, 123, 2643–2653.
- Liefeldt, L.; Schönfelder, G.; Böcker, W.; Hocher, B.; Talsness, C.E.; Rettig, R.; Paul, M. Transgenic rats expressing the human ET-2 gene: A model for the study of endothelin actions in vivo. J. Mol. Med. 1999, 77, 565–574.
- Hocher, B.; Liefeldt, L.; Thöne-Reineke, C.; Orzechowski, H.-D.; Distler, A.; Bauer, C.; Paul, M. Characterization of the Renal Phenotype of Transgenic Rats Expressing the Human Endothelin-2 Gene. Hypertension 1996, 28, 196–201.
- Baynash, A.G.; Hosoda, K.; Giaid, A.; Richardson, J.A.; Emoto, N.; Hammer, R.E.; Yanagisawa, M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 1994, 79, 1277–1285.
- Rice, J.; Doggett, B.; Sweetser, D.A.; Yanagisawa, H.; Yanagisawa, M.; Kapur, R.P. Transgenic rescue of aganglionosis and piebaldism in lethal spotted mice. Dev. Dyn. 2000, 217, 120–132.
- Clouthier, D.E.; Hosoda, K.; Richardson, J.A.; Williams, S.C.; Yanagisawa, H.; Kuwaki, T.; Kumada, M.; Hammer, R.E.; Yanagisawa, M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development 1998, 125, 813–824.
- Yanagisawa, H.; Hammer, R.E.; Richardson, J.A.; Williams, S.C.; Clouthier, D.E.; Yanagisawa, M. Role of Endothelin-1/Endothelin-A receptor-mediated signaling pathway in the aortic arch patterning in mice. J. Clin. Investig. 1998, 102, 22–33.
- Hosoda, K.; Hammer, R.E.; Richardson, J.A.; Baynash, A.G.; Cheung, J.C.; Giaid, A.; Yanagisawa, M. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell 1994, 79, 1267–1276.
- Yanagisawa, H.; Yanagisawa, M.; Kapur, R.P.; Richardson, J.A.; Williams, S.C.; Clouthier, D.E.; de Wit, D.; Emoto, N.; Hammer, R.E. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development 1998, 125, 825–836.
More