Plant lignans exhibit a wide range of biological activities, which makes them the research objects of potential use as therapeutic agents. They provide diverse naturally-occurring pharmacophores and are available for production by chemical synthesis. A large amount of accumulated data indicates that lignans of different structural groups are apt to demonstrate both anti-inflammatory and antioxidant effects, in many cases, simultaneously.
1. Introduction
Through their vital activity, plants produce a wide range of pharmacologically active natural compounds. Phenylpropane (C
6C
3) units, provided by precursor phenylalanine and tyrosine, are found in many natural compounds, including lignans. Lignans are dimer compounds originating from cinnamic acid and its derivatives that also give rise to lignin, pre-eminent polymer component of the plant cell wall. The term “lignans” is often restricted to molecules in which two phenylpropane units are coupled at the central carbon of the side-chain (β-β′-coupling) while compounds with alternative coupling are referred to as neolignans
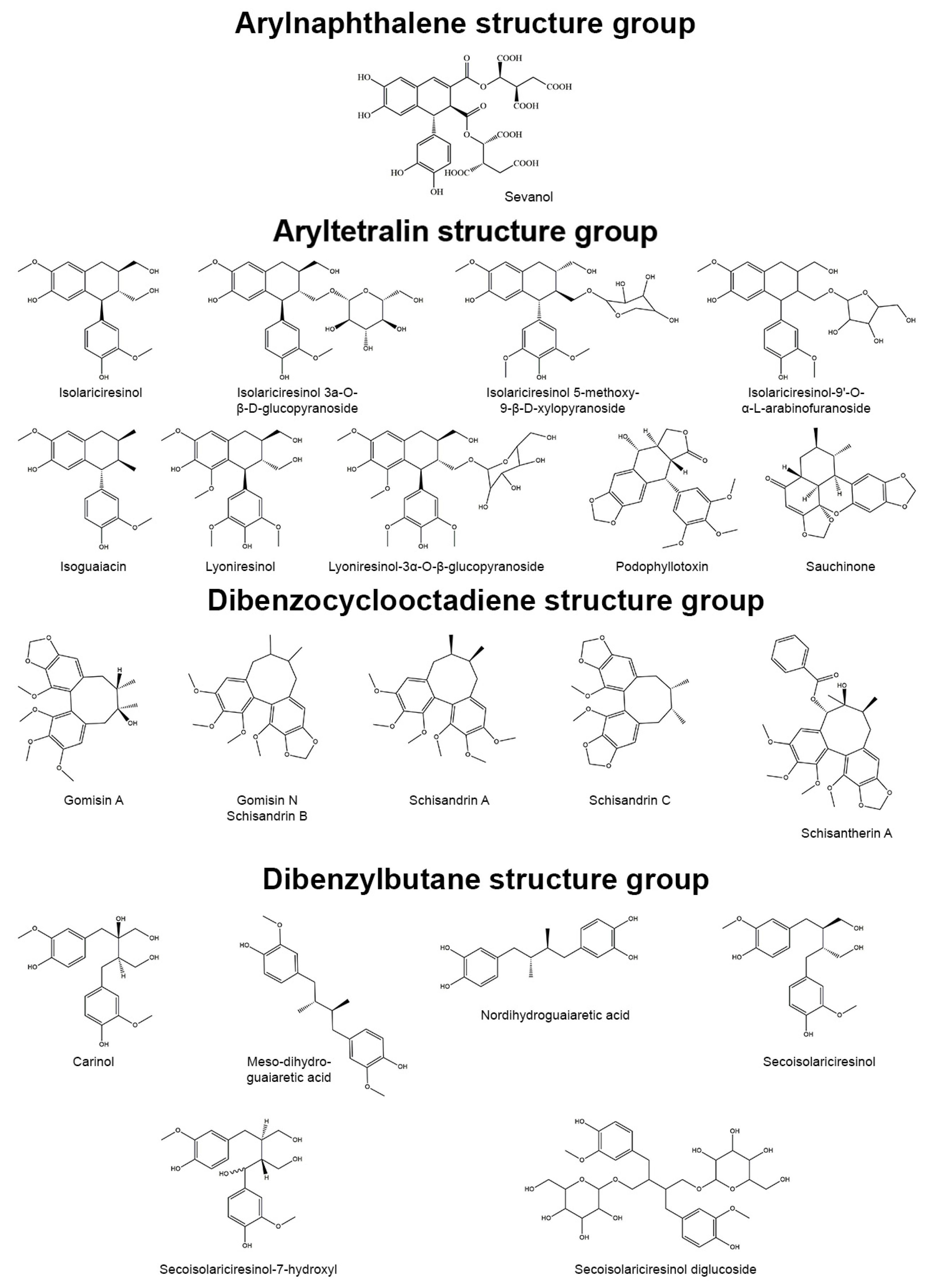
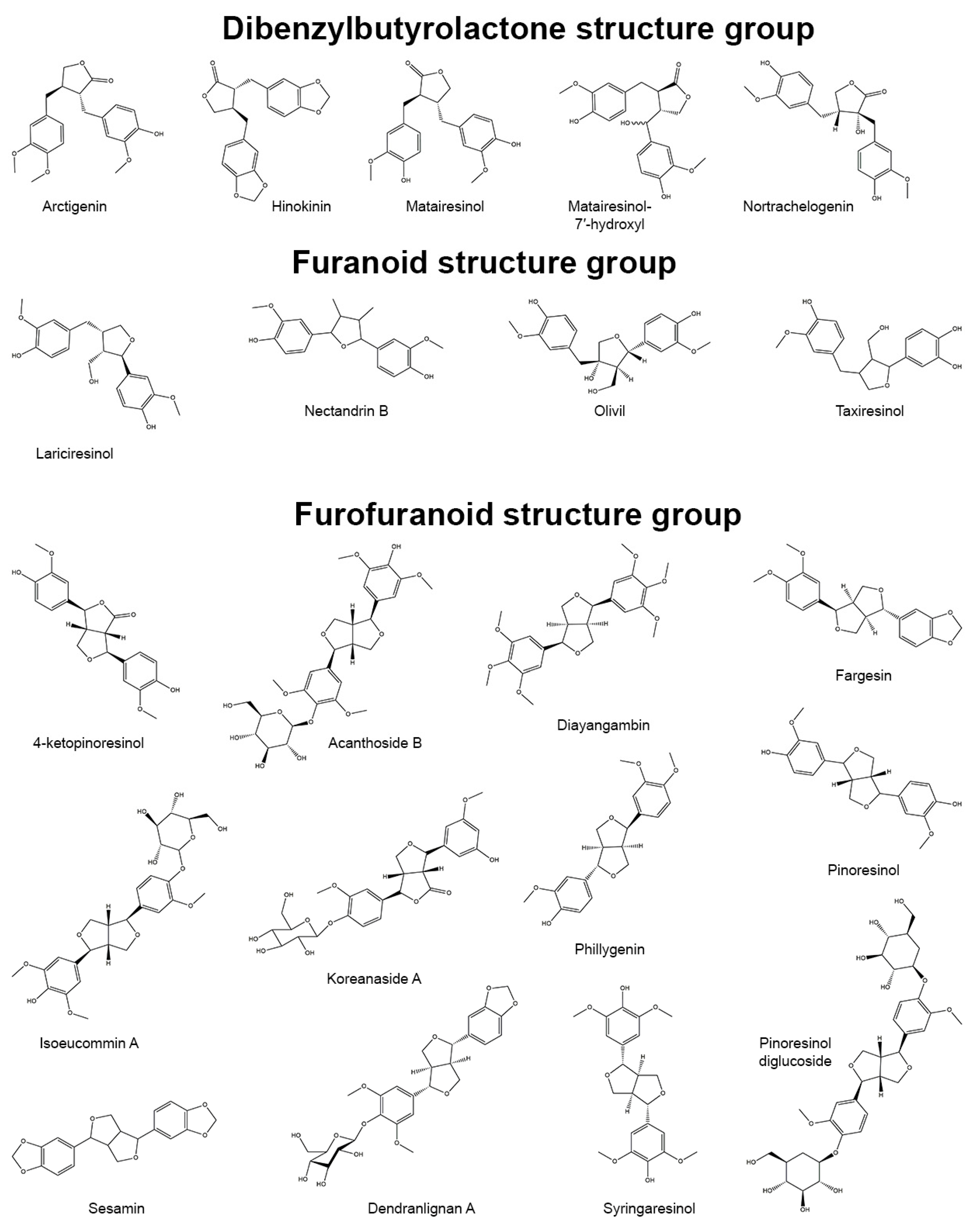
[1]. Based on the patterns of cyclization and oxygen incorporation, lignans can be classified into eight subgroups: arylnaphthalenes, aryltetralins, dibenzocyclooctadienes, dibenzylbutanes, dibenzylbutyrolactones, dibenzylbutyrolactols, furans, and furofurans (
Figure 1). Along with the diversity of structure, lignans exert a broad spectrum of biological activities, e.g., antitumor, antiviral, hepatoprotective, immunosuppressive, anti-platelet, and cardiovascular effects
[2]. Additionally, some lignans can produce strong antioxidant and anti-inflammatory effects.
Figure 1. Representatives of plant lignans of various structural subgroups with antioxidant and anti-inflammatory properties.
Oxidative stress is an imbalance of the oxidants/antioxidants tilting toward an oxidative status, which is characterized by a higher level of reactive oxygen species (ROS) and reactive nitrogen species (RNS) than in the normal physiological state. It could be triggered by heavy metals, xenobiotics, free radicals, drugs, and ionizing radiation. Exposure to these toxicants and oxidants impairs cellular components (e.g., lipids, proteins, and nucleic acids) and initiates the pathogenesis of diabetes mellitus, cancer, neurodegenerative, cardiovascular, lung diseases, etc.
[3].
Inflammation is an adaptive response induced by pathogens, tissue damage or ingestion of allergens or pollutants that includes activation of innate and adaptive immunity. This process is coordinated by a complex regulatory network of factors that fall into four functional categories: inducers, sensors, mediators, and effectors. Endogenous and exogenous inducers activate specialized sensors, e.g., toll-like receptors, inflammasome, IgE, TRP and ASIC channels, etc. They, in turn, elicit the production of specific sets of mediators (vasoactive amines, vasoactive peptides, fragments of complement components, eicosanoids, cytokines, chemokines, and proteolytic enzymes) that alter the functionality of tissues and organs (downstream effectors). Irrespective of injury or infection, long-term stress and malfunction of tissues can also induce chronic low-level inflammation that is common to such disorders as obesity, type 2 diabetes, atherosclerosis, asthma, cancer, autoimmune and neurodegenerative diseases
[4]. The pathological effect of simultaneously developing oxidative stress and inflammation is enormous and requires pharmacological measures for control.
2. Molecular Mechanisms of Inflammation and Oxidative Stress
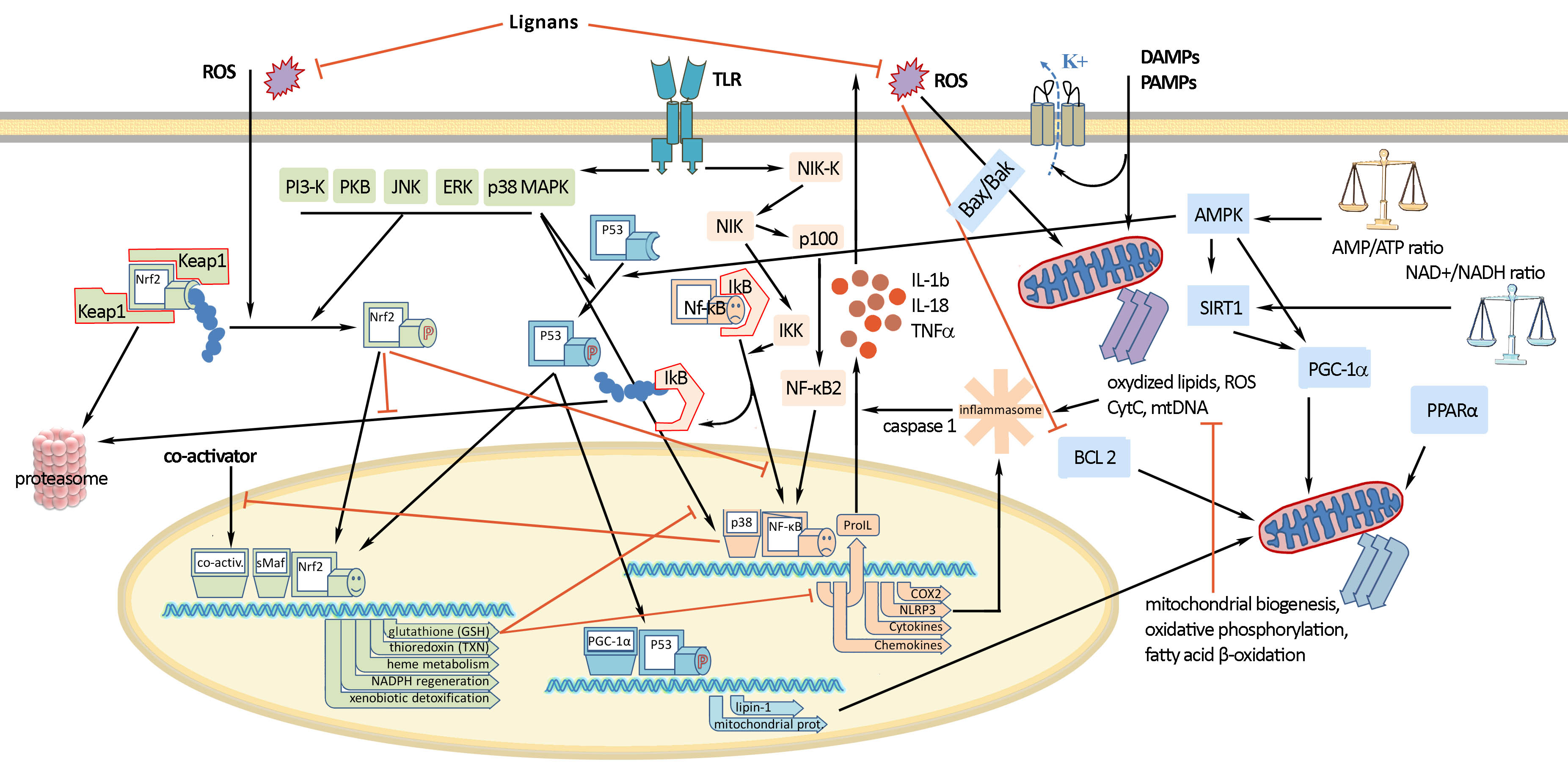
Signals of inflammation can be divided into damage-associated molecular patterns (DAMP), which are cell-derived and initiate immunity in response to trauma and tissue damage, and pathogen-associated molecular patterns (PAMP), which are derived from microorganisms
[5][6]. They stimulate protective reactions via the interaction with pattern recognition receptors with the subsequent formation of inflammasomes. Activation of the inflammasome requires two events: priming and activation. For example, the priming can include recognition of a bacterial lipopolysaccharide (LPS) by toll-like receptor 4 (TLR4) which leads to the activation of a cascade of reactions resulting in the translocation of nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) into the cell nucleus (
Figure 2). Activation of NF-κB is the general event in the inflammatory reaction of a cell and includes two main signaling pathways: canonical and non-canonical (or alternative). The main mechanism of the canonical activation of NF-κB is the induced degradation of the NF-κB-IκB complex triggered by site-specific phosphorylation by IκB kinase (IKK)
[7]. The inhibitory subunit IκBα then leaves the complex and, via ubiquitinylation, is degraded in the proteasome
[8]. Non-canonical activation of NF-κB does not involve IκBα degradation, but depends on the processing of NF-κB2 precursor protein, called p100, by NF-κB-inducing kinase (NIK), which activates and functionally interacts with IKK. The processing of p100 with the degradation of its C-terminal IκB-like structure leads to the formation of a mature NF-κB2 p52 and the nuclear translocation of this non-canonical NF-κB complex into the nucleus
[9]. Canonical or alternatively activated NF-κB in the nucleus promotes the transcription of NF-κB-dependent genes, such as the NLR family pyrin domain containing 3 (NLRP3), pro-Il-1ß and pro-Il-18, which are necessary for the activation of the inflammasome. Mitochondrial damage, as well as plasma membrane damage and K+ efflux, are also considered upstream mechanisms that regulate inflammasome activation
[10]. Finally, activated inflammasome produces inflammatory mediators for excretion, which includes the maturation of pro-inflammatory cytokines such as interleukin-1-beta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α) by caspase-1.
Figure 2. Molecular mechanisms of oxidative stress and inflammation and their interaction. Nuclear factor erythroid-related factor 2 (Nrf2) and nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) are key factors mediating the antioxidant and anti-inflammatory effects of lignans.
Nuclear factor erythroid-related factor 2 (Nrf2) is a redox-sensitive transcription factor that plays an essential role in the protection against oxidative stress and electrophilic injury by regulating a battery of cytoprotective genes. Under basal conditions, Nrf2 binds to its repressor Kelch-like ECH-associated protein 1 (Keap1) and is maintained at a low level in the cytosol through Keap1-mediated ubiquitinylation and 26S proteasome-mediated degradation. The activation of the Nrf2-mediated defensive response is an effective means of counteracting exogenous oxidative insults. Electrophilic and oxidative stressors, such as ROS, can activate Nrf2 promoting its dissociation from Keap1 or phosphorylation by several kinases, including advanced protein kinase B (Akt), extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (p38 MAPK), and protein kinase C (PKC). The activated Nrf2 is guided into the nucleus, forms a heterodimer with a small musculo-aponeurotic fibrosarcoma (Maf) protein, binds to specific DNA sequences called the antioxidant responsive element (ARE) consensus and subsequently initiates the transcription of downstream cytoprotective genes. These ARE-containing genes include various redox-balancing proteins and phase II enzymes, such as heme oxygenase-1 (HO-1) and NAD(P)H:quinone oxidoreductase 1 (NQO1), γ-glutamyl cysteine synthetase (γ-GCS) and glutamate-cysteine ligase (GCL), thioredoxin (Trx), thioredoxin reductase (TrxR) and peroxiredoxin (Prx) as well as glutathione (GSH)-utilizing enzymes, such as superoxide dismutase (SOD), glutathione peroxidase (GPx) and catalase (CAT), glutathione S-transferase (GST), glutathione reductase (GR). These proteins maintain the cellular redox capacity, eliminate ROS, promote excretion of toxicants, and ensure cytoprotection
[11][12][13].
The inflammation and ROS protection cascades regulate each other in several ways (
Figure 2). For example, the Nrf2 transcription factor has been shown to negatively control the NF-κB signaling pathway through various mechanisms. First, Nrf2 inhibits the oxidative stress-mediated activation of NF-κB by reducing intracellular ROS levels
[14]. The activation of Nrf2 has also been reported to inhibit the LPS-induced production of pro-inflammatory cytokines, including IL-6 and IL-1ß, through an ROS-independent mechanism. This was due to the negative regulation of NF-IκB-mediated transcription of pro-inflammatory cytokine genes and genes involved in the inflammasome assembly, such as NLRP3 and caspase 1
[15]. In addition, Nrf2 prevents IκB-α from degradation, thereby inhibiting the nuclear translocation of NF-κB
[16]. NF-κB, in turn, prevents transcriptional co-activators from binding to Nrf2 and subsequent ARE transcription
[17]. It was also found that GSH suppresses the activation of p38, as well as the expression of cyclooxygenase-2 (COX-2) in peritoneal macrophages of rats exposed to LPS stimulation
[18], which shows that the content of GSH can strongly affect the activity and function of molecular and cellular mediators of inflammatory processes.
The mitochondrial status also plays an important role in the regulation of both inflammatory and oxidative stress processes. The degradation of mitochondria through the Bax/Bak pathway leads to an increase in intracellular ROS, oxidized lipids and mtDNA, which provokes the formation of the inflammasome and NF-κB-mediated inflammatory response. Mitochondrial biogenesis, as well as glucose transport and fatty acid oxidation, is improved by the transcription factor PGC-1α. The increase in both AMP/ATP and NAD+/NADH ratio triggers PGC-1α activation through its AMPK-mediated phosphorylation and SIRT1-mediated deacetylation
[19][20]. PGC-1α can also activate Nrf2 by inhibiting the GSK3ß regulator, which prevents the translocation of Nrf2 into the nucleus by phosphorylation. Under oxidative stress, GSK3β is inactivated by p38, which is positively regulated by PGC-1α, and, as a consequence, the antioxidant defense gets activated by Nrf2
[21].
p53 is another protein whose activity depends on the levels of ROS/RNS in oxidative stress conditions. p53 acts as a metabolic regulator or even an apoptosis inducer
[22]. AMPK and p38 MAPK can phosphorylate p53 and induce its interaction with PGC-1α to enhance Nrf2 response and reduce the effects of ROS/RNS
[23].
3. Lignans Action as Antioxidant and Anti-Inflammatory Agents
Numerous data have confirmed the anti-inflammatory and antioxidant activity of lignans both in vitro and in vivo, which are well described in the original review.
Numerous data have confirmed the anti-inflammatory and antioxidant activity of lignans both in vitro and in vivo, which are well summarized in the original review.
4. Main Approaches for Lignan Synthesis
Reviews over the past decades describe a huge variety of different approaches for the racemic or enantioselective synthesis of plant lignans. Overall, the key step is the dimerization of two monomers that are derivatives of coniferyl alcohol. The difference among chemical strategies for obtaining lignans depends on the skeleton peculiaritiesprovided by natural biosynthesis. Wide varieties of elegant methods to obtain these compounds have been already published. Classical pathways include oxidative dimerization, the Diels-Alder reaction, aldol reaction, and the Stobbe condensation
[24][25].