This entryarticle aims to introducereview the physiological roles and pathological implications of oxidative stress in cardiovascular tissues
- reactive oxygen species
- oxidative stress
- antioxidant
- kinase
- mitochondria
- cardiovascular diseases
1. Introduction
Cardiovascular diseases are multifactorial disorders that represent the leading causes of death worldwide according to the World Health Organization (WHO) [1]. The physiopathology of cardiovascular diseases, mainly caused by atherosclerosis, includes remodeling of blood vessels that can result in blood flow restrictions affecting the heart and the nervous system. Cardiovascular diseases comprise several disorders such as coronary artery diseases, stroke, hypertension, heart failure, congenital heart diseases, and vascular diseases. The main risk factors for cardiovascular diseases are obesity, diabetes, cigarette smoking, a sedentary and unhealthy lifestyle, and genetic predisposition [
2. Physiological Roles of Oxidative Stress in Cardiovascular Tissues
Under physiological conditions, low levels of ROS production are equivalent to their detoxification and play a major role in cellular signaling and function [2]. This process is called redox signaling and is defined as the specific and reversible oxidation/reduction modification of cellular signaling components able to regulate gene expression, excitation–contraction coupling, or cell growth, migration, differentiation, and death [3][4] (
Under physiological conditions, low levels of ROS production are equivalent to their detoxification and play a major role in cellular signaling and function [19]. This process is called redox signaling and is defined as the specific and reversible oxidation/reduction modification of cellular signaling components able to regulate gene expression, excitation–contraction coupling, or cell growth, migration, differentiation, and death [20,21] (Figure 1).
).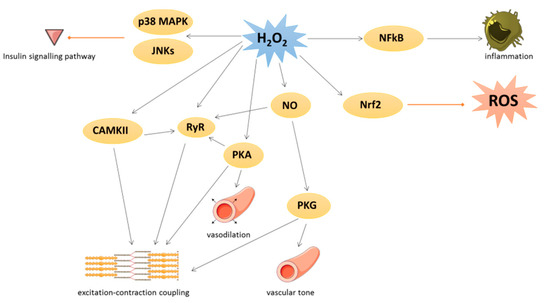
Figure 1.
2
2: hydrogen peroxide; NFκB: nuclear factor-kappa beta; CAMKII: Ca/calmodulin-dependent kinase II; RyR: ryanodine receptor; PKA: cAMP-induced protein kinase A; NO: nitric oxide; PKG: protein kinase G; Nrf2: nuclear factor erythroid 2-related factor 2.
Several kinases are involved in redox signaling. For instance, H
Several kinases are involved in redox signaling. For instance, H2
O2 could activate Ca/calmodulin-dependent kinase II (CAMKII), leading to excitation–contraction coupling [4] or p38 mitogen-activated protein kinase (p38 MAPK) and c-Jun N-terminal kinase (JNKs) leading to inhibition of insulin signal transduction. cAMP-induced protein kinase A (PKA) is also activated by oxidation of its regulatory subunit R1α and translocated from cytosol to membrane, where PKA regulates cardiac excitation–contraction coupling in the heart and vasodilation in the vessels [4].
could activate Ca/calmodulin-dependent kinase II (CAMKII), leading to excitation–contraction coupling [21] or p38 mitogen-activated protein kinase (p38 MAPK) and c-Jun N-terminal kinase (JNKs) leading to inhibition of insulin signal transduction. cAMP-induced protein kinase A (PKA) is also activated by oxidation of its regulatory subunit R1α and translocated from cytosol to membrane, where PKA regulates cardiac excitation–contraction coupling in the heart and vasodilation in the vessels [21].Many transcription factors are also regulated by redox signaling. As an example, the nuclear factor-kappa beta (NFκB) is activated when ROS have damaged its inhibitor (IkB) and regulates inflammation process [5]. The nuclear factor erythroid 2-related factor 2 (Nrf2) could be induced by lipid peroxidation to activate several key antioxidant enzymes containing an antioxidant/electrophile response element motif in their promoter, such as heme oxygenase 1, glutathione peroxidases, SOD, peroxiredoxins, thioredoxins, and thioredoxin reductases [6]. In detail, under unstressed conditions, Nrf2 is constitutively ubiquitinated by both Kelch-k-like ECH-associated protein 1 (Keap1) and Cullin-3 E3 ligase to be damaged [6][7]. The activation of Nrf2 is due to the oxidation of Keap1 that abrogated its negative control on Nrf2.
Many transcription factors are also regulated by redox signaling. As an example, the nuclear factor-kappa beta (NFκB) is activated when ROS have damaged its inhibitor (IkB) and regulates inflammation process [22]. The nuclear factor erythroid 2-related factor 2 (Nrf2) could be induced by lipid peroxidation to activate several key antioxidant enzymes containing an antioxidant/electrophile response element motif in their promoter, such as heme oxygenase 1, glutathione peroxidases, SOD, peroxiredoxins, thioredoxins, and thioredoxin reductases [23]. In detail, under unstressed conditions, Nrf2 is constitutively ubiquitinated by both Kelch-k-like ECH-associated protein 1 (Keap1) and Cullin-3 E3 ligase to be damaged [23,24]. The activation of Nrf2 is due to the oxidation of Keap1 that abrogated its negative control on Nrf2.Under physiological conditions, NO is a cytoprotective molecule with a vasodilator action. NO inhibits the activation and adhesion of platelets and neutrophils and has protective effects against ischemia reperfusion and heart failure [8]. NO could exert its biological effects by binding to the soluble guanylate cyclase in order to produce cyclic guanosine monophosphate leading to protein kinase G (PKG) activation [9] or by S-nitrosylation. This latter modification could modulate several protein activities such as pro-caspase 3, myosin heavy chain, tropomyosin, peroxiredoxins, or ryanodine receptor (RyR). RyR, which mediates Ca
Under physiological conditions, NO is a cytoprotective molecule with a vasodilator action. NO inhibits the activation and adhesion of platelets and neutrophils and has protective effects against ischemia reperfusion and heart failure [19,25]. NO could exert its biological effects by binding to the soluble guanylate cyclase in order to produce cyclic guanosine monophosphate leading to protein kinase G (PKG) activation [26] or by S-nitrosylation. This latter modification could modulate several protein activities such as pro-caspase 3, myosin heavy chain, tropomyosin, peroxiredoxins, or ryanodine receptor (RyR). RyR, which mediates Ca2+ release from sarcoplasmic reticulum, is also activated by phosphorylation via PKA and CAMKII, themselves redox-regulated [4]. Moreover, PKG is activated by oxidation independently of NO and regulates vascular tone and cardiomyocyte contraction or hypertrophy [4].
release from sarcoplasmic reticulum, is also activated by phosphorylation via PKA and CAMKII, themselves redox-regulated [21]. Moreover, PKG is activated by oxidation independently of NO and regulates vascular tone and cardiomyocyte contraction or hypertrophy [21].