Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Lindsay Dong and Version 2 by Mariusz Mojzych.
Metastasis is perhaps the most common reason for treatment failure in cancer patients, as well as the leading cause of cancer-related death. Mitogen-activated protein kinase (MAPKs) are serine/threonine-protein kinases that can be activated by a variety of extracellular stimuli including growth factors, cytokines, insulin, environmental factors, and oxidative and genotoxic stress. It is becoming increasingly clear that MAPKs are involved in all the steps required for hyperproliferating cells to develop into metastatic tumors. However, we are currently lacking in vivo data to fully understand how MAPK signaling pathways can affect the progression of metastatic disease.
- cancer
- c-JUN N-terminal kinase (JNK)
- extracellular signal-regulated kinase (ERK)
- metastasis
- mitogen-activated kinases (MAPKs)
1. Molecular Basis of Metastasis
In all cases, tumor formation starts with the initiating mutation that confers the unlimited proliferative potential of cells, followed by the development of genetic instability that leads to autonomic transformed cells. However, the oncogenic transformation is not sufficient for cells to acquire the ability to enter systemic circulation and infiltrate distant tissues. Moreover, the cells need to survive in the new environment and colonize the foreign tissue. The genes that contribute to all these events can be classified into several classes: (a) metastasis initiation genes, (b) metastasis progression genes, and (c) metastasis virulence genes. The first class of genes (initiation genes) allows tissue invasion and their dispersion. This class includes various factors that contribute to angiogenesis, cell motility, or invasion. Progression genes, on the other hand, facilitate tumor development at the site of metastasis. Similar to oncogenes, progression genes contribute to tumorigenesis; however, during the metastatic process they play additional, advantageous functions that enable the tissue-specific spread. The classical examples of these include MMPs. Metastasis virulence genes are responsible for the aggressive potential of tumor cells not in the primary site, but in the secondary site. It is, however, often difficult to classify a gene to a specific class of metastasis genes as their functions usually overlap [35][1].
So far, hundreds of genes have been shown to contribute to the invasive potential of cells. Mutations in genes encoding the cellular tumor antigen p53 (TP53), cyclin-dependent kinase inhibitor 2A (CDKN2A), phosphatase and tensin homolog (PTEN), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α (PIK3CA), retinoblastoma (RB1), GTPase KRas (KRAS), estrogen receptor (ESR1), MYC proto-oncogene protein (MYC), and serine/threonine-protein kinase B-raf (BRAF) were linked with the tumor metastasis process. However, metastatic research focuses mainly on gene expression changes during the metastatic process, rather than the exploration of specific gene mutations [36][2].
To acquire the abilities needed to form metastases, epithelial stem cells or differentiated epithelial cells must undergo the epithelial–mesenchymal transition (EMT). The whole EMT process is mediated by various transcription factors involved in developmental programs such as twist-related protein 1 (TWIST), zinc finger proteins SNAI1/2 and SLUG, zinc finger E-box-binding homeobox 1/2 (ZEB1/2), forkhead box protein C2 (FOXC2), or paired mesoderm homeobox protein 1 (PRRX1) [37,38][3][4]. The expression levels of noncoding RNAs such as microRNAs and long non-coding RNAs have also been found to change during the EMT process, suggesting that they may play a role in the process [39,40,41,42,43][5][6][7][8][9]. The epigenetic regulation, chromatin remodeling, alternative splicing, posttranslational modifications, stabilization, and altered subcellular localization of proteins all contribute to the EMT [44][10].
Two main mechanisms of cell movement can be distinguished: amoeboid and mesenchymal. The mesenchymal type of movement is dependent on protease (MMPs) activities that enable the movement through the degradation on ECM, while the amoeboid type is a protease-independent process and relies on mechanical forces exerted by the cells on the ECM. Both types of movements are controlled by the signaling pathways of the RHO family of small GTPases that control actin dynamics and their rearrangement during cell migration in response to environmental stimuli. In the mesenchymal type of movement, cells attain a specific elongated spindle-like shape and resemble fibroblasts in shape. RHOA facilitates actin stress fiber formation, whereas Ras-related protein (RAC1) and cell division control protein 42 homolog (CDC42) promote the formation of lamellipodia (network of the actin filaments) and filopodia (rod-like projections of actin fibers) [52][11]. The activity of Rho GTPases is regulated by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs). RHO-associated kinase (ROCK) interacts with formin diaphanous 1 (DIA1) and facilitates actin polymerization. Furthermore, ROCK inactivates myosin-light-chain phosphatase (MLCP), which normally dephosphorylates myosin II light chain (MLC2). The phosphorylation of MLC2 leads to the augmented activity of myosin II ATPase, facilitating its interaction with actin filaments to confer cell contraction. Phosphorylated MLC2 is negatively regulated by myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK). Furthermore, ROCK1 is negatively regulated by RHO-related GTP-binding protein (RHOE). In contrast, 3-phosphoinositide-dependent protein kinase 1 (PDK1) promotes ROCK1-dependent actomyosin filament formation. PI3K is a key regulator of front–rear polarity and is involved in the recruitment of CDC42 and RAC GEFs to the leading edges of the moving cells. RAC and CDC42 regulate actin polymerization via the regulation of Wiskott–Aldrich syndrome protein (WASP) and its interaction with Arp2/3 complex 34 kDa subunit (ARP2/3) [47,52][11][12].
In the amoeboid type of migration, rounded cells move via constant cycles of expansion and contraction of the cell body. This is allowed by actin and myosin, which localize cortically and contribute to membrane blebbing. Blebbing results from the inflow of cytoplasm. In contrast, in lamellipodia, actin polymerization is the underlying cause of changes and movement. Again, the RHO/ROCK signaling pathway regulates the process of migration [6,52][11][13]. In summary, invadopodia, lamellipodia, filopodia, podosomes, and are actin-rich membrane protrusions formed by metastatic cells. To migrate and invade, protrusions use mechanical forces and proteases. They are either the cell’s sensory organelles (filopodia) for signals such as chemoattractants, or the major organelles for cell motility (lamellipodia), or both allowing motility and ECM degradation (invadopodia). Unlike single-cell migration, collectively migrating cells maintain cell–cell connections by continuously expressing adhesion molecules. Cells may travel as sheets, strands, tubes (coordinated invasion), or clusters (cohort migration). Collective cell migration involves force creation to pull or push cells forward or backward. Substrate-binding integrins generate the energy required for motility. Integrins are expressed on the leading edges of cells to form adhesion complexes with ECM components such as fibronectin. ECM attachment stimulates cytoskeletal adaptor proteins, such as cortactin, vinculin, paxillin, and talin. Migrating cells develop membrane protrusions and integrin-mediated focal adhesions connected to the actin cytoskeleton. Cells also express MMPs at their leading edges to split collagen fibers and arrange them in tube-like structures to travel in the cell mass [6][13].
MMPs constitute a family of 24 endopeptidases that regulate the ECM composition through proteolytic activity. Based on their structure and function, MMPs can be classified into eight groups encompassing MMPs either secreted or membrane-bound. Due to their proteolytic function, the activity of MMPs must be tightly controlled. This is achieved through strictly controlled transcriptional and translational events. Moreover, MMPs are expressed as proenzymes or zymogens that need to be activated by “cysteine switch” mechanisms and autocatalysis reactions in which the enzyme cleaves its prodomain and gains catalytic activity. Furthermore, as previously mentioned, the activity of enzymes is controlled by four tissue inhibitors of metalloproteinases (TIMPs) that impede their enzymatic function [53][14]. Due to their proteolytic activity, MMPs have been suggested to play a critical role in all of the metastatic steps from the invasion through ECM degradation [54,55[15][16][17],56], angiogenesis [54,57[15][18][19],58], immune evasion [59,60[20][21][22][23],61,62], the establishment of premetastatic niche [63,64][24][25], extravasation [65][26], to proliferation and survival in the new environment [66,67,68][27][28][29]. Thus, the inhibition of MMPs may represent a new strategy in targeting metastasis. Despite the obvious roles of MMPs in tumor metastasis, continuous efforts fail to implement MMPs inhibitors into the clinic. This is attributed to the difference between murine and human biology, e.g., differences in lifespan, differences in tumor growth and size without metastatic spread, or the use of genetically homogenous cancer cells in the bolus injection compared with the greater heterogenicity of tumor cells in humans. These factors, together with the lack of the specificity of inhibitors and the potential side-effects, have restricted the use of MMP inhibitors in cancer treatment. Moreover, MMPs act early in the development of metastasis; thus, certain actions, such as MMP expression profiling, should be performed in the pre-and peri-metastatic stages. Both antibodies and small-molecule inhibitors targeting MMPs with varied affinity, specificity, and selectivity have been developed [53][14].
2. The MAPK Pathway in Metastasis
MAPKs are serine/threonine-protein kinases that can be activated by a variety of extracellular stimuli including growth factors, cytokines, insulin, environmental factors, and oxidative and genotoxic stress. Through the use of genetically engineered mouse models and chemically induced tumorigenesis models it has been observed that components of the MAPK pathway not only regulate the behavior of tumor cells, but also the behavior of surrounding normal stromal cells in the TME during cancer pathogenesis. The unique activities of MAPK pathway components in tumor initiation and development vary based on the stimuli and stromal cells involved in tumor growth, as well as the molecular isoforms of the pathway components, as reviewed in [153,154][30][31]. The conventional MAPKs in mammals include c-Jun NH2-terminal kinase (JNK), P38 MAPK, and extracellular signal-regulated kinase (ERK), which, in turn, exist in several isoforms. JNK1 and JNK2 are found in nearly all tissues in contrast to JNK3, which is found primarily in neuronal cells. P38 MAPK exists in several isoforms encompassing P38α (also known as MAPK14 or SAPK2a), P38β (MAPK11, SAPK2b), P38γ (MAPK12, SAPK3, ERK6), and P38δ (MAPK13, SAPK4). ERK1 and ERK2 are the subtypes of the eight isoforms of ERK that are activated by MAPK/ERK kinase 1 (MEK1/2) [155,156][32][33]. In contrast, ERK3/4 and ERK7/8 are considered atypical MAPKs [157][34]. The MAPK pathway components are shown in Figure 41.
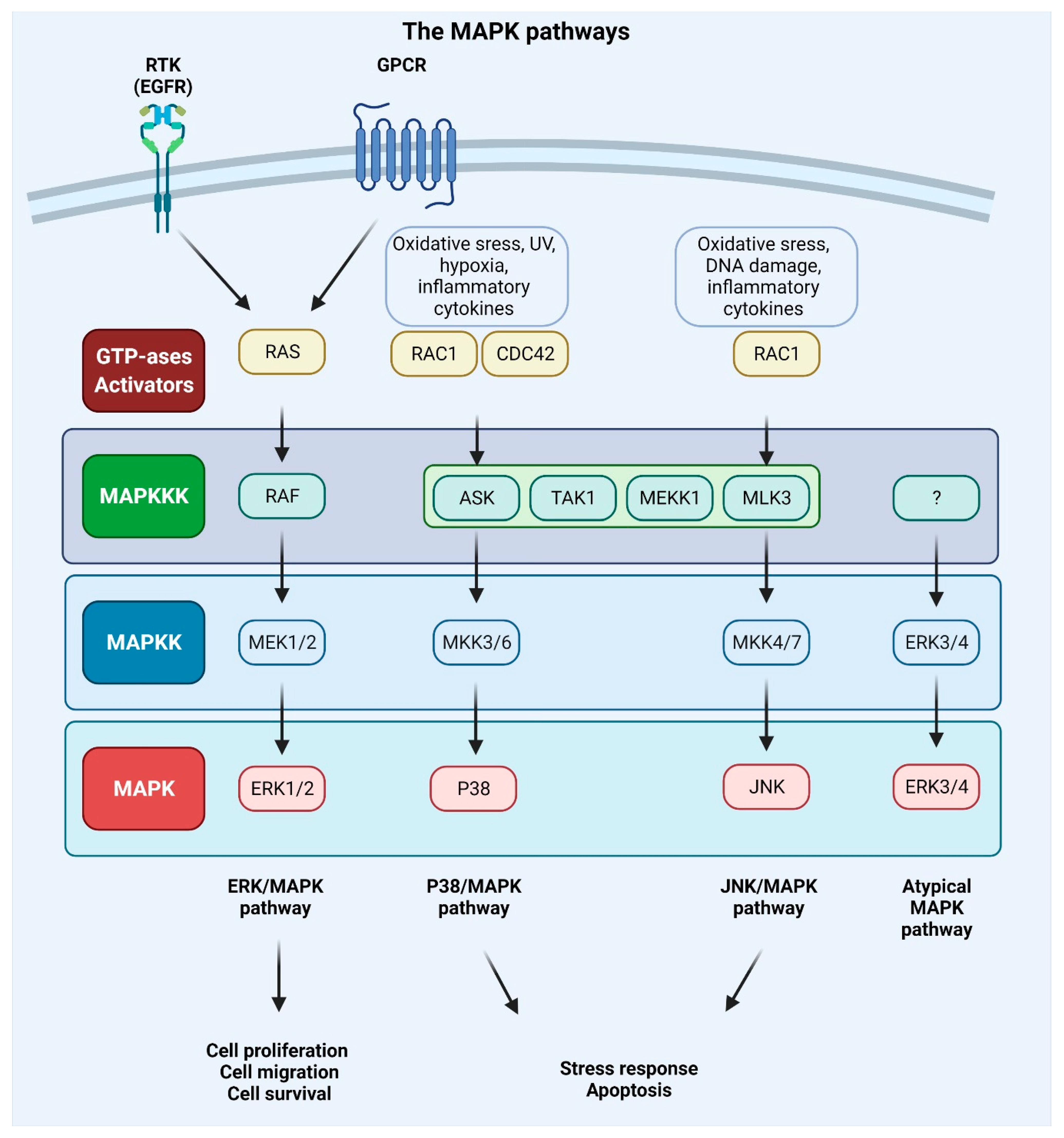
Figure 41. Mitogen-activated protein kinase (MAPK) signaling pathways. Extracellular signals such as growth factors and cytokines, as well as intracellular signals such as oxidative and DNA damage, activate MAPK pathways. GTPases (activators) such as RAS, RAS-related protein (RAC), and cell division control protein 42 homolog (CDC42) constitute the first layer of MAPK signaling cascade, which conveys the signal to downstream protein kinases. The MAPK signaling cascades consist of three kinases: mitogen-activated protein kinase kinase kinase (MAPKKK), a mitogen-activated protein kinase kinase (MAPKK), and mitogen-activated protein kinase (MAPK) and results in proliferation, migration, differentiation, survival, or apoptosis. Mammalian MAPK pathways include ERK MAPK, P38 MAPK, and JNK MAPK signaling events. ERK/MAPK pathway is activated by RAS, which is attracted to the plasma membrane through receptor tyrosine kinases (RTKs) and G protein-coupled receptor (GPCRs) activation. In this cascade, MAPK/ERK kinase 1/2 (MEK1/2) activates extracellular signal-regulated kinase 1/2 (ERK1/2). P38/MAPK and JNK/MAPK pathways are triggered by various insults that activate signaling through MKK3/6 or MKK4/7 (MAPKKs), respectively, that are activated upon MAPKKK s such apoptosis signal-regulating kinase 1 (ASK1), transforming growth factor-β-activated kinase 1 (TAK1), MEKK1 (MAPKKK), and MLK3 (MAPKKK). ERK3/4 are considered atypical MAPK kinases [156][33].
Every MAPK signaling cascade involves at least three core kinases: MAPKKK (mitogen-activated protein kinase kinase kinase), MAPKK (mitogen-activated protein kinase kinase), and MAPK (mitogen-activated protein kinase) [158][35]. MAPK pathways are present in nearly all eukaryotes and play an important role in numerous cellular activities, including gene expression, metabolism, proliferation, apoptosis, invasion, and metastasis [156][33]. The RAS–RAF–MEK–ERK pathway is disrupted in approximately 40% of all human malignancies, with mutations in BRAF (10%) and its upstream activator RAS (30%) being the most frequently observed. Inhibitors of MEK were among the first anti-MAPK pathway therapeutics to be created, but despite their high potency and selectivity, they failed in clinical trials. This was due to the negative feedback mechanisms of the pathway components and systemic toxicity of the drugs. Treating metastatic malignant melanoma and other cancers with the use of a combination of RAF and MEK inhibitors has become standard practice. The majority of the pioneering work has been performed in metastatic malignant melanoma, which is characterized by a high prevalence of BRAF (50–60%) and NRAS (15–20%) mutations [159,160,161,162,163][36][37][38][39][40].
The MAPK pathway is initiated by the binding of growth factor to the RTK or GPCR and the subsequent phosphorylation of RAS protein and following activation of BRAF or RAF (also known as MAPKKK) kinase. The activation signal is conveyed to the MAPKKs (phosphorylation of two serine residues of the MEK1 or MEK2 protein). The downstream phosphorylation of tyrosine and threonine residues of ERK kinase results in the phosphorylation of a multitude of protein substrates involved in differentiation, apoptosis, and migration [41,156][7][33]. Upon stimulation of MAPK signaling, ERK1/2 shuttles from cytoplasm to the nucleus, where it regulates gene expression by phosphorylating numerous transcription factors. In the cytoplasm, cytoskeletal components such as microtubule-associated protein (MAP1, MAP2, MAP4) are the targets of the ERK1/2 kinase. These phosphorylation events control the cell morphology and cytoskeletal redistribution. Moreover, ERK1/2 may phosphorylate other cytoplasmic components, including son of sevenless (SOS), RAF1, and MEK, providing a negative feedback regulation of the pathway. In the nucleus, proto-oncogenes including c-FOS, c-JUN, ETS domain-containing protein (ELK1), c-MYC, and cyclic AMP-dependent transcription factor (ATF2) are all phosphorylated in an ERK1/2-dependent manner [126][41]. The activation of the ERK/MAPK signaling pathway can promote tumor invasion and metastasis by upregulating MMP expression, whereas the inhibition of this signaling can impede the aforementioned processes [164,165][42][43]. It was also discovered that mesothelin regulates the expression of MMP7 through the MAPK/ERK signal transduction pathway, as well as the ERK1/2, AKT, and JNK-mediated pathways, contributing to the invasiveness of ovarian cancer cells [166][44]. Moreover, RAS-associated protein RAP1A was identified as a significant promoter of ovarian cancer cell metastasis via activation of ERK and P38 signaling and the induction of EMT through enhanced expression of SLUG, ZEB1, vimentin, fibronectin, and MMP9 [167][45].
The modification of cellular adhesiveness has a direct impact on the mobility of cells. The activation of the ERK/MAPK pathway has been demonstrated to regulate the disassembly of focal adhesions [158][35]. Moreover, fibroblast de-adhesion triggered by EGFR necessitates the activation of M-calpain, which is downstream of the ERK/MAPK kinase signaling pathway [168,169,170,171][46][47][48][49]. Previous studies indicate that activation of the MAPK pathway may not be sufficient for the induction of cell mobility and may require phospholipase C activity (PLC) [172,173][50][51]. Moreover, the RAS/RAF/MEK/ERK kinase cascade can have a profound impact on HIF-1α protein translation. Activated ERK phosphorylates eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), ribosomal protein S6 kinase (S6K), and MAP kinase interacting kinase (MNK) (which can, in turn, directly phosphorylate eukaryotic translation initiation factor 4E (eIF-4E)) and enhances mRNA translation of HIF-1α protein involved in the response of cells to hypoxia [174][52]. Moreover, ERK may alter MMP activity, which affects gastric cancer (GC) cell migration or invasion, and many proteins upstream of the ERK/MAPK pathway, such as IL-22, RasGAP-activating-like protein 1 (RASAL1), protein tyrosine phosphatase type IVA 3 (PRL3), nuclear apoptosis-inducing factor 1 (NAIF1), coiled-coil domain-containing protein 134 (CCDC134), and zinc finger protein (ZIC1) that potentially affect invasion and migration in GC cell lines [158][35]. The RAS/RAF/ERK cell signaling pathway and the P38 MAPK pathway are both responsible for the activation of MNKs that are engaged in oncogenic transformation and can promote metastasis. Alternative splicing results in the production of four MNKs isoforms in human cells (MNK1a/b and MNK2a/b), which are derived from two genes. Through the regulation of eukaryotic translation initiation factor 4E (eIF4E), these kinases play a critical role in the control of the expression of specific proteins involved in the cell cycle, cell survival, and cell motility. However, they also regulate the expression of genes through the modulation of other substrates such as heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), polypyridine tract-binding protein-associated splicing factor (SFPQ), and sprouty 2 (SPRY2)[53]. This topic was recently reviewed in [175].
ROS play a critical role in the regulation of various biological processes. ROS are an integral part of the tumor microenvironment and may promote cancer angiogenesis, metastasis, and survival. Several studies have demonstrated that ROS accumulation is a significant contributor to the EMT process and this topic has been previously reviewed [176,177,178][54][55][56]. For example, ROS causes epigenetic alterations in the promoter region of E-cadherin and several other tumor suppressor genes, resulting in tumor development and metastasis. It may cause gene promotor hypermethylation via SNAI-mediated induction of histone deacetylase 1 (HDAC1) and DNA methyltransferase 1 (DNMT1). Moreover, it was found that TGF-β1 controls the expression of uPA and MMP9, which aids in cell motility and invasion through ROS-mediated events. ROS present in moderate concentrations, stimulate the activation of the cancer cell survival signaling cascade, which includes the MAPK/ERK, P38, JNK, and PI3K/AKT signaling. As a result of the pathway activation ROS contribute to the activation of NF-κB, MMPs, and VEGF. However, cells have to maintain a balance between ROS generation and elimination, as excess ROS production may lead to DNA damage and apoptosis [125][57]. Moreover, EGFR/RAS/MAPK signaling pathway is involved in NFκB activation, cyclooxygenase-2 (COX2) upregulation, and GC cell proliferation. COX2 upregulation promotes cancer growth and decreases apoptosis.
The P38 pathway includes the MAPKKKs such as apoptosis signal-regulating kinase 1 (ASK1), transforming growth factor-β-activated kinase 1 (TAK1), mitogen-activated protein kinase kinase kinase 1 (MEKK1), and mixed-lineage kinase 3 (MLK3), and MAPKKs, such as MKK3/6, which in turn activate P38 [70][58]. In various malignancies, P38 promotes EMT and metastasis via the upregulation of pro-metastatic genes [179,180][59][60]. P38 MAPKs perform a wide range of functions through binding to and activating a diverse array of substrates. More than 100 proteins have been demonstrated to be susceptible to direct phosphorylation by P38 MAPKs in vitro and in vivo, with approximately half of these being transcription factors, including ATF-1, -2, and -6, TP53, and CCAAT/enhancer-binding protein α (C/EBPα). Other substrates include protein kinases (e.g., MAP kinase-activated protein kinase 2/3 (MK2/3), ribosomal protein S6 kinase α 5 (MSK1) and phosphatases (e.g., serine/threonine-protein phosphatase 2A catalytic subunit α isoform (PPP2CA)), cell-cycle proteins (e.g., cyclin D1), apoptosis proteins (e.g., BCL-2 family proteins), growth factor receptors (e.g., fibroblast growth factor receptor 1 (FGFR1)), and cytoskeletal proteins (e.g., tau, keratin 8) [181][61]. For more information on substrates of P38 see [182,183][62][63]; moreover the table, a companion to the SnapShot “p38 MAPK Signaling” in the January 31 issue of Cell, describes 66 P38α substrates grouped into eight different categories based on biochemical function [184][64].
It has been demonstrated that P38 may be involved in the phosphorylation of the Ser68 residue on TWIST1, which leads to increased protein stability and promotes its capacity to induce EMT and invasiveness in breast cancer [185][65]. It has been found that elevated TWIST1 levels are also dependent on activation of the ERK signaling [186][66]. TWIST1 may in turn act as a transcriptional factor for MMPs [187][67]. Additionally, P38-mediated signaling was shown to regulate the expression of MMP1, MMP2, MMP9, and MMP13 in multiple cancer cell lines [188,189,190,191,192][68][69][70][71][72]. In contrast, high expression levels of SNAI together with high expression levels of the phosphorylated P38 MAPK (Thr180/Tyr182) were found in primary tumors. High expression of SNAI in metastatic cells is correlated with an increased risk of death in ovarian cancer patients [193][73]. A strong link between inflammation and EMT has been established [194][74]. SNAI was found to trigger IL-6 production, which may in turn act as an EMT trigger. IL-6 also contributes to signal transducer and activator of transcription 3 (STAT3) activation, which affects both tumorigenesis and metastasis [195,196,197][75][76][77]. IL-1β is another cytokine linked to the advancement of cancer including gastric adenocarcinoma, but its molecular causes remain unknown. Both P38 and JNK regulate the IL-1β signaling pathway and the activation of P38 by IL-1β enhances GC cell motility, invasion, and metastatic potential in vitro and in vivo. It was shown that IL-1β induces the IL-1β/P38/AP-1(c-FOS)/MMP2/MMP9 pathway [198][78].
The Forkhead box (FOX) family of transcription factors, which are distinguished by a conserved DNA-binding domain known as the ‘forkhead’ or ‘winged-helix’, regulate a wide range of biological functions, including cell proliferation, differentiation, apoptosis, and metabolism. FOXC1 and FOXC2, play a critical role in the regulation of embryonic, ocular, and cardiac development. A wide variety of cancers are including breast carcinomas, hepatocellular carcinomas, lymphomas exhibit elevated expression of FOXC1 and FOXC2. FOXC transcription factors aid in the progression of cancer through regulation of cell proliferation, metastasis, EMT, and angiogenesis [199][79]. FOXC1 promotes tumor metastasis in numerous human malignant cancers. However, the upstream and downstream molecular mechanisms of FOXC1 in metastasis remain unknown. FOXC1 upregulation was related to poor prognosis in colorectal cancer (CRC). In vitro and in vivo, FOXC1 knockdown reduced CRC cell migration and invasion while FOXC1 overexpression increased the metastatic potential of the tested cells. Moreover, it was found that in metastatic CRC cells, FOXC1 regulates MMP10 and the expression of transcription factors SOX4 and SOX13. FOXC1’s Ser241 and Ser272 were found to be important sites for the interaction with P38, phosphorylation of which contribute to its stability [200][80]. Moreover, the P38-mediated phosphorylation of Ser367 of FOXC2 serves as a regulatory mechanism of ZEB1 in metastatic breast cancer cells. The inhibition of P38–FOXC2 signaling selectively reduces cell metastasis without an effect on primary tumor growth. The genetic or pharmacological suppression of P38 reverses the EMT in a FOXC2-dependent process [201][81].
As described above, ZEB1 was identified as a downstream target of FOXC2 [201][81]. ZEB1 is a transcription factor that belongs to the ZEB family of transcription factors. It is distinguished by the presence of two zinc finger clusters, which are important for DNA binding, as well as a homeodomain that is centrally positioned. Other protein binding domains found in ZEB1 include the Smad interaction domain (SID), the CtBP interaction domain (CID), and the p300-P/CAF binding domain, among others (CBD). ZEB1 can bind to certain DNA sequences known as E-boxes and either downregulate or upregulate the expression of its target gene by recruiting co-suppressors or co-activators through the CID, SID, or CBD signaling pathways. The suppression of ZEB1 in MDA-MB-231 human breast cancer cells results in the overexpression of around 200 genes and the downregulation of approximately 30 genes, the majority of which are determinants of epithelial differentiation and cell–cell adhesion. Because of the critical function of ZEB1 in the downregulation of E-cadherin, it is thought to operate as a driver of the EMT and cancer progression. In addition to suppressing the expression of E-cadherin, ZEB1 regulates the expression of several additional target genes that are implicated in tumor growth. For instance, ZEB1 binds to the promoters of epithelial polarity genes and suppresses their transcription, causing breast cancer cells to lose adherence and thus conferring invasive potential [202][82].
P38 can activate HIF-1 by stabilizing its α subunit (HIF-1α). HIF-1 is also a transcriptional regulator of growth factors and cytokines such as VEGF and TGF-β that are involved in EMT. In addition, HIF-1 can directly stimulate the production of SNAI and TWIST affecting cell migration and EMT [203,204][83][84]. P38α also may trigger cell migration or cytoskeletal remodeling via the phosphorylation of heat-shock protein 27 (HSP27), the activation of LIM domain kinase 1 (LIMK1), and the inactivation of cofilin [205,206][85][86]. Cofilin is a small abundant protein that binds both G-actin (monomeric) and F-actin (filamentous actin) and thus confers cell migration. Several studies have found that the expression of specific genes in the cofilin pathway is altered in invasive tumor cells, suggesting that cofilin is involved in the initiation of the early phases of the motility cycle. Moreover, the cofilin pathway responds to the TME stimuli that are implicated in cell migration through the activation of other pathways (see Section 5) involved in metastasis. These include cytokines and growth factors such as EGF and TGFα [207][87].
P38 responds to ROS buildup by encouraging growth stagnation and death, hence preventing carcinogenesis. TNF-α can be stimulated by ROS, resulting in the activation of the JNK signaling pathway and the induction of apoptosis. On the other hand, TNF-α may activate NF-κB and decrease ROS production through the induction of associated genes such as manganese superoxide dismutase (MnSOD) and ferritin heavy chain (FHC), blocking JNK activation and apoptosis. The activation of P38 stimulates the activity of ribosomal protein S6 kinase α 5/4 (RPS6KA5/4 or MSK1/2) and, in turn, promotes the activity of NF-κB [70][58]. More recently, plasma membrane Ca2+ pump isoform 4b (PMCA4b or ATP2B4) has been established as a metastasis suppressor in BRAF mutant melanoma cells. The activation of P38 triggers the endo/lysosomal internalization and degradation of the ion pump in melanoma cells. Moreover, the inhibition of the P38 MAPK pathway reduces both migration, and metastasis of BRAF mutant cells via the increase in PMCA4b expression and a reduction in β4 integrin yields [208][88].
While other isoforms of P38 were shown to have a profound influence on cancer metastasis, for many years P38δ was a poorly investigated member of the MAPK family. However, it was found that this isoform is highly expressed in particularly all types of human breast cancers, and the inhibition of P38δ in MCF-7 and MDA-MB-231 breast cancer cell lines results in diminished cell proliferation. Moreover, cells without P38δ seem to exhibit enhanced cell–matrix adhesion. This is attributed to the regulatory role of P38δ on FAK kinase [209][89]. Moreover, P38δ was shown to enhance the development of CSCs in breast cancer [210][90]. In contrast, P38γ and P38δ activation may suppress CSCs development in non-small-cell lung cancer (NSCLC) through promotion of the ubiquitin-mediated degradation of SOX2, OCT4, NANOG, KLF4 and MYC transcription factors that normally contribute to the acquisition of cancer stem cell characteristics [211][91]. The role of P38 signaling in metastasis was also previously summarized in [180][60] and is shown in Figure 52.
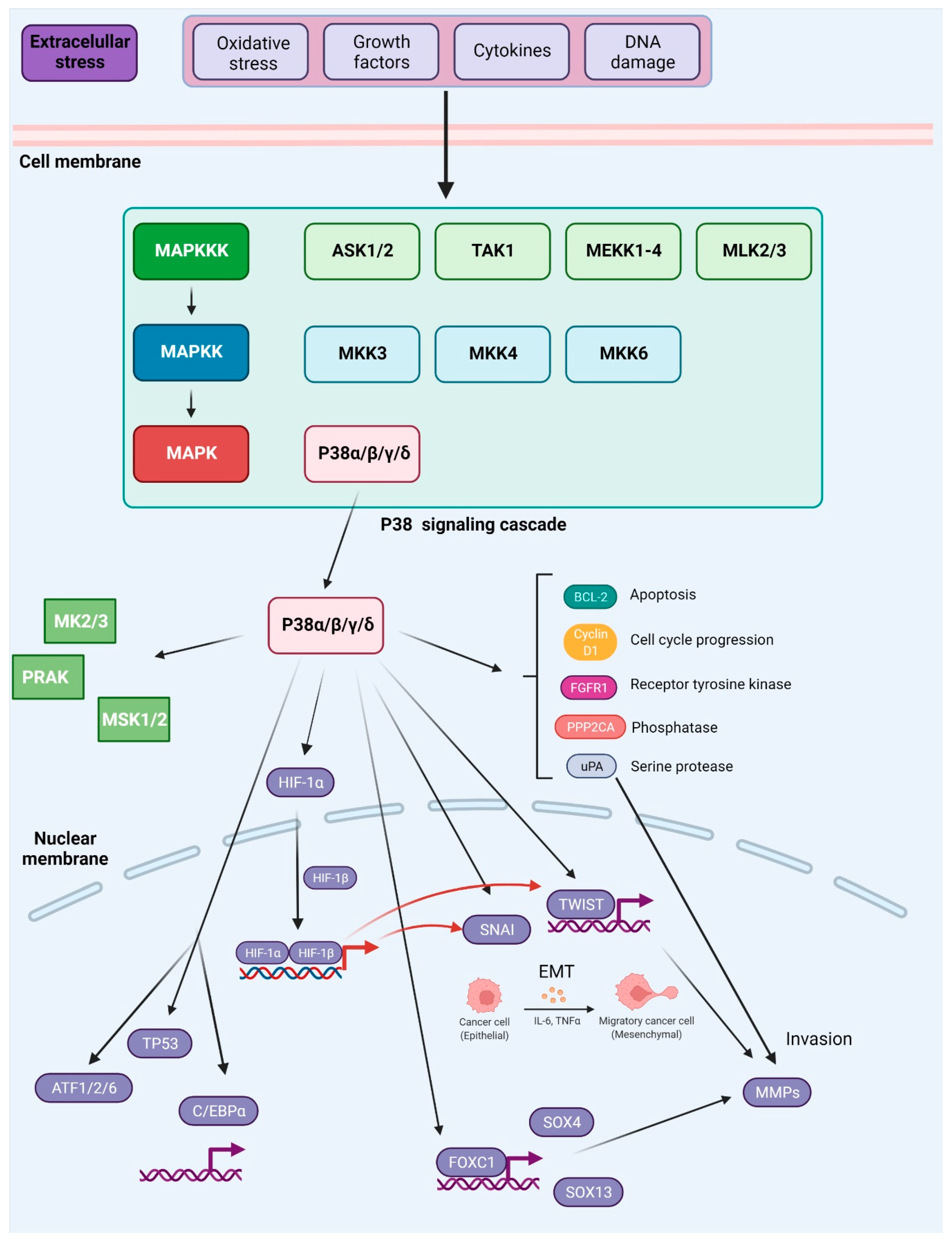
Figure 52. Activation and downstream targets of P38 MAPK. P38 MAPK is activated through several mechanisms. The canonical MAPK signaling module involves sequential phosphorylation and activation events that pass down from MAP3Ks to MAP2Ks, and from MAP2Ks to P38 MAPK. In response to various external stresses and signals (e.g., oxidative stress, UV irradiation, DNA-damage chemotherapeutic agents, and cytokines), several MAP3Ks can trigger activation of P38 signaling, such as TAK1, MEKK1-4, MLK2/3, and ASK1/2. Three MAP2Ks, namely MKK3, MKK6, and MKK4, are direct upstream activators of P38 MAPK. In addition to canonical activation, P38a, the best-characterized member of the P38 kinase family, can also be activated through autophosphorylation. P38 MAPK has been reported to phosphorylate more than 100 proteins, highlighting the versatility of this signaling pathway. Prominent downstream targets include transcription factors, protein kinases, and phosphatases, growth factor receptors, as well as key regulators of cell cycle and apoptosis (depicted in the main text of the article). Based on [181][61].
JNK1, JNK2, and JNK3 are the kinases encoded by genes belonging to the JNK family. JNK1 and JNK2 are expressed throughout the body, whereas JNK3 expression is restricted to certain tissues, with the highest levels found in the brain, heart, and testes. For each of the genes, several different splice variants result in a total of 10 isoforms of the protein with molecular weights ranging from 46 to 54 kDa [212][92]. MKK4 and MKK7 are two representatives of the MAPKK kinases belonging to the JNK sub-pathway activated when MAPKKKs are triggered. These components then phosphorylate and activate JNK, which in turn phosphorylates a multitude of substrates of the AP-1 transcription factor, with c-JUN, FOS, and FOS-related antigen 1/2 (FRA1/2) being the most relevant. Other JNK’s downstream targets include members of the mitochondrial apoptosis regulator BCL-2 family (BCL-2, BCL-xL, BAD, BIM, and BAX), as well as ATF2, ELK-1, TP53, and c-MYC [213,214][93][94].
JNKs have a dualistic role in cancer [215][95]. For example, in mouse embryonic fibroblasts (MEFs), the deletion of JNK2 results in increased cell proliferation, whereas the loss of JNK1 has the opposing effect [216][96]. The differential regulation of c-JUN is thought to be responsible for these effects [217][97]. In MEFs, JNK loss in combination with the double knockout of TP53 (TP53–/–) results in MET, as demonstrated by increased E-cadherin expression, decreased N-cadherin expression, and lower colony-forming ability [218][98]. TGFβ activates JNKs in a cascade that necessitates the involvement of TAK1. This pathway is critical for TGFβ signaling because it is required for the phosphorylation of SMAD3 by JNK and is required for the subsequent transcriptional activation of SMAD3. Not only does the phosphorylation of SMAD3 by JNK increase the efficacy of SMAD-dependent gene expression, but it also increases SMAD3 translocation to the nucleus. Because SMAD3 directly transactivates SNAI1 and SNAI2, JNKs may promote the EMT [212][92].
Furthermore, the role of JNKs in inflammation is well established [219,220,221][99][100][101]. Activated JNK1 promotes the recruitment of inflammatory macrophages, which release VEGF to stimulate angiogenesis and MMPs to aid in tissue remodeling. Moreover, monocytes release TGF-β, which in turn causes tumor cells to undergo the EMT [212][92]. Studies suggest that the double knockout JNK1−/− results in reduction in tumor burden, tumor proliferation, and cytokine production, including TNFα and IL-6. Several studies have suggested that the JNK-dependent inflammatory response promotes tumor progression through induction of the EMT in cells [222][102]. Moreover, JNK-stimulated binding of c-JUN to the VEGF promoter may increase the expression of angiogenic factors facilitating the access of tumor cells to oxygen and nutrients [223,224][103][104]. Phosphorylated JNK activates c-JUN, which results in an increase in the expression of MMP2 as a result of the upregulation of astrocyte elevated gene-1 (AEG-1) in cells. The upregulation of AEG-1 dramatically increases the aggressiveness of osteosarcoma cells via the JNK/c-JUN/MMP2 pathway. In addition, it has been shown that the JNK pathway can promote cancer invasion and metastasis by boosting the expression of other MMP family members such as MMP7 and MMP9, which are induced by the activation of the downstream signaling cascade [213][93].
In human cancer, it is common to observe apparent defects in cell polarity. The fundamental processes through which cell polarity disturbance contributes to tumor growth and metastasis are uncertain. When different apicobasal polarity genes in Drosophila are mutated, JNK signaling is activated and the E-cadherin/β-catenin adhesion complex is downregulated. Both of these events are required and sufficient to cause oncogenic RAS(V12)-induced benign tumors in the developing eye to exhibit metastatic behavior. Furthermore, when oncogenic RAS is present, active JNK and RAS signaling work together to promote tumor development, with JNK signaling switching from a proapoptotic to a pro-growth function depending on the context [225][105].
The overexpression of glucose-regulated protein 94 (GRP94) has been observed in a variety of malignancies, including breast, liver, lung, colorectal, gastric, pancreatic, and head and neck cancers. GRP94 is a key protein involved in mediating cancer progression, and it is highly expressed in hepatocellular carcinoma (HCC). On the other hand, chaperonin-containing TCP1 complex (CCT1-8) proteins are highly conserved molecular chaperones that are involved in promoting the correct folding of newly synthesized proteins or the refolding of misfolded proteins. Furthermore, it has been proposed that CCT proteins are implicated in the progression of a variety of cancers, including breast cancer, colorectal cancer, uterine sarcoma, and lung cancer, among others. CCT8 overexpression has been discovered in a variety of cancers, including colon cancer, breast cancer, glioma, and HCC. It has been reported that the silencing of GRP94 hindered the wound healing, migration, and invasion of HCC cells. These findings suggested that GRP94 knockdown may have a suppressive impact on HCC cell metastasis via a reduction in CCT8/c-JUN/EMT signaling in HCC cells. The silencing of GRP94 greatly reduced the migration and invasion of cells [226][106].
JNK is a multifunctional protein that can mediate both cell transformation and apoptosis through a variety of mechanisms that partially overlap with those of the ERK signaling pathway. JNK has been shown to increase resistance to ERK pathway inhibitors as well as chemotherapeutic agents. Moreover, JNK is unquestionably significant in the development of resistance to RAF inhibitors [159][36]. Evidence suggests that nuclear apoptosis-inducing factor 1 (NAIF1), a protein often downregulated or lost in cancer regulates cellular migration and invasion through the MAPK pathway. The human NAIF1 gene encodes a 327-amino acid protein with a homeodomain-like region and two nuclear localization signals at its N-terminus. The overexpression of NAIF leads to cell growth inhibition and apoptosis. GC cell growth, migration, and invasiveness can be suppressed by NAIF1. NAIF1 can decrease the expression MMP2 and MMP9, and reduce the activity of FAK. Additionally, NAIF1 restrains MAPK1 and MAPK8 activity via the inhibition of their mRNA expression with accompanied ERK and JNK degradation. Thus, the therapeutic targeting of NAIF1 seems to be a new potential strategy in GC treatment [227,228][107][108]. Moreover, JNK1 contributes to the survival of circulating cancer cells via inhibition of the transcription of apoptosis-inhibiting genes. As a result, JNK1 and JNK2 may work in concert to improve CTC survival by boosting survival signals and inhibiting apoptosis [212][92].
Transgelin is an actin-binding protein that is involved in the promotion of cell motility in healthy cells. Although there is debate over whether or not transgelin plays a role in cancer development, many studies have demonstrated that elevated transgelin levels are associated with aggressive tumor behavior, advanced stage of the disease, and poor prognosis [229,230][109][110]. Changes in the expression of the transgelin protein mediated by the AKT and JNK signaling pathways increase the metastatic potential of CRC cells. The suppression of transgelin, AKT, or JNK signaling results in a significant reduction in cell migration and invasion in SW620 cells with the concurrent inhibition of actin cytoskeleton dynamics [231][111].
It has been observed that tenascin-C (TNC), an extracellular matrix glycoprotein, may influence metastases and contribute to the poor prognosis of patients with pancreatic cancer. TNC was shown to induce the migration and invasion of pancreatic cancer cells. This was associated with the upregulation of EMT-associated markers, including MMP9, in a JNK/c-JUN-dependent manner. Moreover, because TNC can activate JNK, it can enhance the association of paxillin with FAK, which promotes pancreatic cancer cell motility and adhesion [232][112]. The role of JNKs in metastasis was summarized elsewhere [212][92].
Several investigations have demonstrated that JNK is involved in the migration and invasion of prostate cancer cells. In PC3 and DU145 cells, the inhibition of JNK pathways by the JNK inhibitor SP600125 or JNK siRNA prevented thrombospondin-2-induced migration and invasion [233][113]. It has been also demonstrated that the CC chemokine receptor 7 (CCR7) may significantly boost the expression of phosphorylated JNK in PC3 cells by activating NOTCH signaling. This results in increased migration and enhanced metastatic activity in PC3 cells [234][114].
ERK signaling events are tightly controlled cascades. These regulatory components include bispecific phosphatases, scaffold proteins, control of signal duration, and intensity, as well as the dynamic subcellular localization of cascade components in response to environmental stimuli [126][41]. More recently, MLK3 has been identified as a crucial player in MAPK signaling with an impact on cell invasion and metastasis. MLK3 belongs to the class of MAPKKK that transduce signals from cell surface receptors to JNK, ERK, and P38 kinases. In mammals, MLK comprises four members: MLK1 (MAP3K9), MLK2 (MAP3K10), MLK3 (MAP3K11), and MLK4 with two isoforms (MLK4α and MLK4β). It is essential for migrating cells to undergo cytoskeletal rearrangement and FA changes, which are controlled both spatially and temporally by the activities of the GTPases CDC42, RAC1, and RHOA. MLK3 works as a scaffold protein for RAF1 and allows subsequent BRAF phosphorylation and activation of MEK1/2 and ERK. MLK3 acts as a negative regulator of RHOA GTPase via direct binding to RHOA-specific guanine exchange factor P63RHO-GEF [235][115]. In breast cancer cells, the catalytic activity of MLK3 is essential for the activation of JNK, which in turn phosphorylates Ser178 of paxillin, resulting in the proliferation of the cancer cells. This phosphorylation event on paxillin engages FAKs, which in turn stimulates further phosphorylation of paxillin on Tyr31 and Tyr118 [236][116]. Phosphorylated paxillin is capable of competing with the RHOA-specific GAP protein, P190RHO-GAP, for binding to the P120RAS-GAP. In this way, when paxillin attaches to P120RAS-GAP, it releases P190RHO-GAP from the binding site of P120RAS-GAP, allowing P190RHO-GAP to decrease the activity of RHOA [237][117]. In addition to being essential for optimal JNK activation. MLK3 distribution in the centrosome and on microtubules during mitosis appears to govern microtubule structure in a JNK-independent manner [238][118]. Several members of the JNK-interacting proteins (JIPs), including JIP-1, -2, and 3, have been shown to function as scaffold proteins for the MLK3-MKK7-JNK signaling subsystem. It has been demonstrated that the JIP-2 protein serves as a docking site for the recruitment of MLK3, MKK3, and either the P38α or P38δ isoforms of MAPK, allowing for MLK3-dependent P38 MAPK activation to occur more efficiently. MLK3 has been demonstrated to signal through a variety of receptors, including EGFR [45][119], and the discoidin domain receptor 1 (DDR1) [83][120]. As a result, downstream JNK, ERK, or P38 signaling is triggered. MLK3 has been also shown to be involved in the invasion of triple-negative breast cancer (TNBC) cells triggered by C-X-C chemokine receptor type 4 (CXCR4)/stromal cell-derived factor 1 (CXCL12). Highly metastatic TNBC cells can be prevented from migrating by inhibiting either the MLK3 or JNK pathways, or by silencing the MLK3 gene. In highly invasive breast cancer cells, the depletion of MLK3 or suppression of its activity leads to increased RHOA activity, excessive FA and stress fiber production, and as a result, reduced cell motility. One possible mechanism by which MLK3 may govern cancer cell invasion is through the regulation of the expression of MMPs. For example, the expression of MMP2 and MMP9 is dependent on the MLK3–ERK–AP1 axis. This suggests that MLK3 may promote cancer invasion in part by upregulating MMPs. Furthermore, MLK3 promotes the EMT switch triggered by collagen type I in prostate cancer. In this model, MLK3 transduces signaling from two collagen receptors, the integrin 2 and the DDR1 receptors, increasing the production of the EMT marker N-cadherin in a process mediated through the MKK7-JNK pathway [239][121]. The function of MLK3 in proliferation, invasion, and metastasis was summareviewized in [239][121].
References
- Nguyen, D.X.; Massagué, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 2007, 8, 341–352.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28.
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890.
- Zhang, J.; Ma, L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012, 31, 653–662.
- Xu, Q.; Deng, F.; Qin, Y.; Zhao, Z.; Wu, Z.; Xing, Z.; Ji, A.; Wang, Q.J. Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis. Cell Death Dis. 2016, 7, e2254.
- Asl, E.R.; Amini, M.; Najafi, S.; Mansoori, B.; Mokhtarzadeh, A.; Mohammadi, A.; Lotfinejad, P.; Bagheri, M.; Shirjang, S.; Lotfi, Z.; et al. Interplay between MAPK/ERK signaling pathway and MicroRNAs: A crucial mechanism regulating cancer cell metabolism and tumor progression. Life Sci. 2021, 278, 119499.
- Magnelli, L.; Schiavone, N.; Staderini, F.; Biagioni, A.; Papucci, L. MAP Kinases Pathways in Gastric Cancer. Int. J. Mol. Sci. 2020, 21, 2893.
- Soleimani, A.; Rahmani, F.; Saeedi, N.; Ghaffarian, R.; Khazaei, M.; Ferns, G.A.; Avan, A.; Hassanian, S.M. The potential role of regulatory microRNAs of RAS/MAPK signaling pathway in the pathogenesis of colorectal cancer. J. Cell. Biochem. 2019, 120, 19245–19253.
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18.
- Paňková, K.; Rösel, D.; Novotný, M.; Brábek, J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 2010, 67, 63–71.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418.
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155.
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015, 44, 94–112.
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183.
- Amano, S.; Akutsu, N.; Matsunaga, Y.; Nishiyama, T.; Champliaud, M.F.; Burgeson, R.E.; Adachi, E. Importance of balance between extracellular matrix synthesis and degradation in basement membrane formation. Exp. Cell Res. 2001, 271, 249–262.
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285.
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370.
- McQuibban, G.A.; Gong, J.H.; Tam, E.M.; McCulloch, C.A.; Clark-Lewis, I.; Overall, C.M. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 2000, 289, 1202–1206.
- Godefroy, E.; Manches, O.; Dréno, B.; Hochman, T.; Rolnitzky, L.; Labarrière, N.; Guilloux, Y.; Goldberg, J.; Jotereau, F.; Bhardwaj, N. Matrix metalloproteinase-2 conditions human dendritic cells to prime inflammatory T(H)2 cells via an IL-12- and OX40L-dependent pathway. Cancer Cell 2011, 19, 333–346.
- Sheu, B.C.; Hsu, S.M.; Ho, H.N.; Lien, H.C.; Huang, S.C.; Lin, R.H. A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 2001, 61, 237–242.
- Guedez, L.; Jensen-Taubman, S.; Bourboulia, D.; Kwityn, C.J.; Wei, B.; Caterina, J.; Stetler-Stevenson, W.G. TIMP-2 targets tumor-associated myeloid suppressor cells with effects in cancer immune dysfunction and angiogenesis. J. Immunother. 2012, 35, 502–512.
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in Cancer Progression and Metastasis. Matrix Biol. 2015, 44, 200–206.
- Rucci, N.; Sanità, P.; Angelucci, A. Roles of metalloproteases in metastatic niche. Curr. Mol. Med. 2011, 11, 609–622.
- Voura, E.B.; English, J.L.; Yu, H.-Y.E.; Ho, A.T.; Subarsky, P.; Hill, R.P.; Hojilla, C.V.; Khokha, R. Proteolysis during Tumor Cell Extravasation In Vitro: Metalloproteinase Involvement across Tumor Cell Types. PLoS ONE 2013, 8, e78413.
- Strand, S.; Vollmer, P.; van den Abeelen, L.; Gottfried, D.; Alla, V.; Heid, H.; Kuball, J.; Theobald, M.; Galle, P.R.; Strand, D. Cleavage of CD95 by matrix metalloproteinase-7 induces apoptosis resistance in tumour cells. Oncogene 2004, 23, 3732–3736.
- Newby, A.C. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc. Res. 2006, 69, 614–624.
- George, S.J.; Dwivedi, A. MMPs, cadherins, and cell proliferation. Trends Cardiovasc. Med. 2004, 14, 100–105.
- Kamiyama, M.; Naguro, I.; Ichijo, H. In vivo gene manipulation reveals the impact of stress-responsive MAPK pathways on tumor progression. Cancer Sci. 2015, 106, 785–796.
- Wortzel, I.; Seger, R. The ERK Cascade. Genes Cancer 2011, 2, 195–209.
- Kim, E.K.; Choi, E.-J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882.
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83.
- Olea-Flores, M.; Zuñiga-Eulogio, M.D.; Mendoza-Catalán, M.A.; Rodríguez-Ruiz, H.A.; Castañeda-Saucedo, E.; Ortuño-Pineda, C.; Padilla-Benavides, T.; Navarro-Tito, N. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial–Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 2885.
- Yang, M.; Huang, C.-Z. Mitogen-activated protein kinase signaling pathway and invasion and metastasis of gastric cancer. World J. Gastroenterol. 2015, 21, 11673–11679.
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102.
- Fiore, M.; Forli, S.; Manetti, F. Targeting Mitogen-activated Protein Kinase-activated Protein Kinase 2 (MAPKAPK2, MK2): Medicinal Chemistry Efforts to Lead Small Molecule Inhibitors to Clinical Trials. J. Med. Chem. 2016, 59, 3609–3634.
- Kiniwa, Y.; Okuyama, R. Recent advances in molecular targeted therapy for unresectable and metastatic BRAF-mutated melanoma. Jpn. J. Clin. Oncol. 2021, 51, 315–320.
- Djanani, A.; Eller, S.; Öfner, D.; Troppmair, J.; Maglione, M. The Role of BRAF in Metastatic Colorectal Carcinoma–Past, Present, and Future. Int. J. Mol. Sci. 2020, 21, 9001.
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, E2801.
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007.
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, E1618.
- Gao, J.; Wang, Y.; Yang, J.; Zhang, W.; Meng, K.; Sun, Y.; Li, Y.; He, Q.-Y. RNF128 Promotes Invasion and Metastasis Via the EGFR/MAPK/MMP-2 Pathway in Esophageal Squamous Cell Carcinoma. Cancers 2019, 11, E840.
- Chang, M.-C.; Chen, C.-A.; Chen, P.-J.; Chiang, Y.-C.; Chen, Y.-L.; Mao, T.-L.; Lin, H.-W.; Lin Chiang, W.-H.; Cheng, W.-F. Mesothelin enhances invasion of ovarian cancer by inducing MMP-7 through MAPK/ERK and JNK pathways. Biochem. J. 2012, 442, 293–302.
- Lu, L.; Wang, J.; Wu, Y.; Wan, P.; Yang, G. Rap1A promotes ovarian cancer metastasis via activation of ERK/p38 and notch signaling. Cancer Med. 2016, 5, 3544–3554.
- Glading, A.; Chang, P.; Lauffenburger, D.A.; Wells, A. Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J. Biol. Chem. 2000, 275, 2390–2398.
- Glading, A.; Uberall, F.; Keyse, S.M.; Lauffenburger, D.A.; Wells, A. Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J. Biol. Chem. 2001, 276, 23341–23348.
- Glading, A.; Lauffenburger, D.A.; Wells, A. Cutting to the chase: Calpain proteases in cell motility. Trends Cell Biol. 2002, 12, 46–54.
- Glading, A.; Bodnar, R.J.; Reynolds, I.J.; Shiraha, H.; Satish, L.; Potter, D.A.; Blair, H.C.; Wells, A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol. Cell. Biol. 2004, 24, 2499–2512.
- Chen, P.; Xie, H.; Sekar, M.C.; Gupta, K.; Wells, A. Epidermal growth factor receptor-mediated cell motility: Phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J. Cell Biol. 1994, 127, 847–857.
- Chen, P.; Murphy-Ullrich, J.E.; Wells, A. A role for gelsolin in actuating epidermal growth factor receptor-mediated cell motility. J. Cell Biol. 1996, 134, 689–698.
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389.
- Pinto-Díez, C.; Ferreras-Martín, R.; Carrión-Marchante, R.; González, V.M.; Martín, M.E. Deeping in the Role of the MAP-Kinases Interacting Kinases (MNKs) in Cancer. Int. J. Mol. Sci. 2020, 21, 2967.
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial-mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036.
- Wang, Z.; Li, Y.; Sarkar, F.H. Signaling Mechanism(S) of Reactive Oxygen Species in Epithelial-Mesenchymal Transition Reminiscent of Cancer Stem Cells in Tumor Progression. Curr. Stem Cell Res. Ther. 2010, 5, 74–80.
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Reactive oxygen species induce epithelial-mesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx-2/Snail signaling pathways in MCF-7 cells. Mol. Med. Rep. 2019, 20, 2339–2346.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Chen, C.; Nelson, L.J.; Ávila, M.A.; Cubero, F.J. Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link. Cells 2019, 8, 1172.
- Wu, X.; Zhang, W.; Font-Burgada, J.; Palmer, T.; Hamil, A.S.; Biswas, S.K.; Poidinger, M.; Borcherding, N.; Xie, Q.; Ellies, L.G.; et al. Ubiquitin-conjugating enzyme Ubc13 controls breast cancer metastasis through a TAK1-p38 MAP kinase cascade. Proc. Natl. Acad. Sci. USA 2014, 111, 13870–13875.
- del Barco Barrantes, I.; Nebreda, A.R. Roles of p38 MAPKs in invasion and metastasis. Biochem. Soc. Trans. 2012, 40, 79–84.
- Zou, X.; Blank, M. Targeting p38 MAP kinase signaling in cancer through post-translational modifications. Cancer Lett. 2017, 384, 19–26.
- Whitmarsh, A.J. A central role for p38 MAPK in the early transcriptional response to stress. BMC Biol. 2010, 8, 47.
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417.
- Trempolec, N.; Dave-Coll, N.; Nebreda, A.R. SnapShot: p38 MAPK substrates. Cell 2013, 152, 924.
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990.
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.-P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89.
- Weiss, M.B.; Abel, E.V.; Mayberry, M.M.; Basile, K.J.; Berger, A.C.; Aplin, A.E. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer Res. 2012, 72, 6382–6392.
- Hsieh, M.-J.; Chen, K.-S.; Chiou, H.-L.; Hsieh, Y.-S. Carbonic anhydrase XII promotes invasion and migration ability of MDA-MB-231 breast cancer cells through the p38 MAPK signaling pathway. Eur. J. Cell Biol. 2010, 89, 598–606.
- Johansson, N.; Ala-aho, R.; Uitto, V.; Grénman, R.; Fusenig, N.E.; López-Otín, C.; Kähäri, V.M. Expression of collagenase-3 (MMP-13) and collagenase-1 (MMP-1) by transformed keratinocytes is dependent on the activity of p38 mitogen-activated protein kinase. J. Cell Sci. 2000, 113, 227–235.
- Kumar, P.; Yadav, A.; Patel, S.N.; Islam, M.; Pan, Q.; Merajver, S.D.; Teknos, T.N. Tetrathiomolybdate inhibits head and neck cancer metastasis by decreasing tumor cell motility, invasiveness and by promoting tumor cell anoikis. Mol. Cancer 2010, 9, 206.
- Park, S.Y.; Jeong, K.J.; Panupinthu, N.; Yu, S.; Lee, J.; Han, J.W.; Kim, J.M.; Lee, J.-S.; Kang, J.; Park, C.G.; et al. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene 2011, 30, 1351–1359.
- Xu, L.; Chen, S.; Bergan, R.C. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 2006, 25, 2987–2998.
- Hipp, S.; Berg, D.; Ergin, B.; Schuster, T.; Hapfelmeier, A.; Walch, A.; Avril, S.; Schmalfeldt, B.; Höfler, H.; Becker, K.-F. Interaction of Snail and p38 mitogen-activated protein kinase results in shorter overall survival of ovarian cancer patients. Virchows Arch. 2010, 457, 705–713.
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075.
- Zhou, J.; Zhang, C.; Pan, J.; Chen, L.; Qi, S.-T. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human adamantinomatous craniopharyngioma cells and promotes tumor cell migration. Mol. Med. Rep. 2017, 15, 4123–4131.
- Sun, Q.; Shang, Y.; Sun, F.; Dong, X.; Niu, J.; Li, F. Interleukin-6 Promotes Epithelial-Mesenchymal Transition and Cell Invasion through Integrin β6 Upregulation in Colorectal Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 8032187.
- Lyons, J.G.; Patel, V.; Roue, N.C.; Fok, S.Y.; Soon, L.L.; Halliday, G.M.; Gutkind, J.S. Snail up-regulates pro-inflammatory mediators and inhibits differentiation in oral keratinocytes. Cancer Res. 2008, 68, 4525–4530.
- Huang, Q.; Lan, F.; Wang, X.; Yu, Y.; Ouyang, X.; Zheng, F.; Han, J.; Lin, Y.; Xie, Y.; Xie, F.; et al. IL-1β-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol. Cancer 2014, 13, 18.
- Chen, X.; Wei, H.; Li, J.; Liang, X.; Dai, S.; Jiang, L.; Guo, M.; Qu, L.; Chen, Z.; Chen, L.; et al. Structural basis for DNA recognition by FOXC2. Nucleic Acids Res. 2019, 47, 3752–3764.
- Zhang, Y.; Liao, Y.; Chen, C.; Sun, W.; Sun, X.; Liu, Y.; Xu, E.; Lai, M.; Zhang, H. p38-regulated FOXC1 stability is required for colorectal cancer metastasis. J. Pathol. 2020, 250, 217–230.
- Werden, S.J.; Sphyris, N.; Sarkar, T.R.; Paranjape, A.N.; LaBaff, A.M.; Taube, J.H.; Hollier, B.G.; Ramirez-Peña, E.Q.; Soundararajan, R.; den Hollander, P.; et al. Phosphorylation of serine 367 of FOXC2 by p38 regulates ZEB1 and breast cancer metastasis, without impacting primary tumor growth. Oncogene 2016, 35, 5977–5988.
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487.
- Emerling, B.M.; Platanias, L.C.; Black, E.; Nebreda, A.R.; Davis, R.J.; Chandel, N.S. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell. Biol. 2005, 25, 4853–4862.
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314.
- Laferriere, J.; Houle, F.; Taher, M.M.; Valerie, K.; Huot, J. Transendothelial migration of colon carcinoma cells requires expression of E-selectin by endothelial cells and activation of stress-activated protein kinase-2 (SAPK2/p38) in the tumor cells. J. Biol. Chem. 2001, 276, 33762–33772.
- Kobayashi, M.; Nishita, M.; Mishima, T.; Ohashi, K.; Mizuno, K. MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006, 25, 713–726.
- Wang, W.; Eddy, R.; Condeelis, J. The cofilin pathway in breast cancer invasion and metastasis. Nat. Rev. Cancer 2007, 7, 429–440.
- Naffa, R.; Vogel, L.; Hegedűs, L.; Pászty, K.; Tóth, S.; Kelemen, K.; Singh, N.; Reményi, A.; Kállay, E.; Cserepes, M.; et al. P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b. Cells 2020, 9, E1209.
- Wada, M.; Canals, D.; Adada, M.; Coant, N.; Salama, M.F.; Helke, K.L.; Arthur, J.S.; Shroyer, K.R.; Kitatani, K.; Obeid, L.M.; et al. P38 delta MAPK promotes breast cancer progression and lung metastasis by enhancing cell proliferation and cell detachment. Oncogene 2017, 36, 6649–6657.
- Xu, M.; Wang, S.; Wang, Y.; Wu, H.; Frank, J.A.; Zhang, Z.; Luo, J. Role of p38γ MAPK in regulation of EMT and cancer stem cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3605–3617.
- Fang, Y.; Wang, J.; Wang, G.; Zhou, C.; Wang, P.; Zhao, S.; Zhao, S.; Huang, S.; Su, W.; Jiang, P.; et al. Inactivation of p38 MAPK contributes to stem cell-like properties of non-small cell lung cancer. Oncotarget 2017, 8, 26702–26717.
- Ebelt, N.D.; Cantrell, M.A.; Van Den Berg, C.L. c-Jun N-Terminal Kinases Mediate a Wide Range of Targets in the Metastatic Cascade. Genes Cancer 2013, 4, 378–387.
- Li, Y.-S.; Deng, Z.-H.; Zeng, C.; Lei, G.-H. JNK pathway in osteosarcoma: Pathogenesis and therapeutics. J. Recept. Signal Transduct. Res. 2016, 36, 465–470.
- Tam, S.Y.; Law, H.K.-W. JNK in Tumor Microenvironment: Present Findings and Challenges in Clinical Translation. Cancers 2021, 13, 2196.
- Tournier, C. The 2 Faces of JNK Signaling in Cancer. Genes Cancer 2013, 4, 397–400.
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 2004, 15, 713–725.
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400.
- Cellurale, C.; Sabio, G.; Kennedy, N.J.; Das, M.; Barlow, M.; Sandy, P.; Jacks, T.; Davis, R.J. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol. Cell. Biol. 2011, 31, 1565–1576.
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, E857.
- Guma, M.; Firestein, G.S. c-Jun N-Terminal Kinase in Inflammation and Rheumatic Diseases. Open Rheumatol. J. 2012, 6, 220–231.
- Kaminska, B. Molecular characterization of inflammation-induced JNK/c-Jun signaling pathway in connection with tumorigenesis. Methods Mol. Biol. 2009, 512, 249–264.
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97.
- Krejsgaard, T.; Vetter-Kauczok, C.S.; Woetmann, A.; Lovato, P.; Labuda, T.; Eriksen, K.W.; Zhang, Q.; Becker, J.C.; Ødum, N. Jak3- and JNK-dependent vascular endothelial growth factor expression in cutaneous T-cell lymphoma. Leukemia 2006, 20, 1759–1766.
- Saníger, M.L.; Oya, R.; Macías, D.; Domínguez, J.N.; Aránega, A.; Luque, F. c-Jun kinase mediates expression of VEGF induced at transcriptional level by Rac1 and Cdc42Hs but not by RhoA. J. Cell. Biochem. 2006, 98, 650–660.
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146.
- Wei, P.-L.; Huang, C.-Y.; Tai, C.-J.; Batzorig, U.; Cheng, W.-L.; Hunag, M.-T.; Chang, Y.-J. Glucose-regulated protein 94 mediates metastasis by CCT8 and the JNK pathway in hepatocellular carcinoma. Tumour Biol. 2016, 37, 8219–8227.
- Yang, M.; Gu, Y.-Y.; Peng, H.; Zhao, M.; Wang, J.; Huang, S.-K.; Yuan, X.-H.; Li, J.; Sang, J.-L.; Luo, Q.; et al. NAIF1 inhibits gastric cancer cells migration and invasion via the MAPK pathways. J. Cancer Res. Clin. Oncol. 2015, 141, 1037–1047.
- Lv, B.; Shi, T.; Wang, X.; Song, Q.; Zhang, Y.; Shen, Y.; Ma, D.; Lou, Y. Overexpression of the novel human gene, nuclear apoptosis-inducing factor 1, induces apoptosis. Int. J. Biochem. Cell Biol. 2006, 38, 671–683.
- Zhou, H.-M.; Fang, Y.-Y.; Weinberger, P.M.; Ding, L.-L.; Cowell, J.K.; Hudson, F.Z.; Ren, M.; Lee, J.R.; Chen, Q.-K.; Su, H.; et al. Transgelin increases metastatic potential of colorectal cancer cells in vivo and alters expression of genes involved in cell motility. BMC Cancer 2016, 16, 55.
- Lin, Y.; Buckhaults, P.J.; Lee, J.R.; Xiong, H.; Farrell, C.; Podolsky, R.H.; Schade, R.R.; Dynan, W.S. Association of the actin-binding protein transgelin with lymph node metastasis in human colorectal cancer. Neoplasia 2009, 11, 864–873.
- Zhou, H.; Zhang, Y.; Chen, Q.; Lin, Y. AKT and JNK Signaling Pathways Increase the Metastatic Potential of Colorectal Cancer Cells by Altering Transgelin Expression. Dig. Dis. Sci. 2016, 61, 1091–1097.
- Cai, J.; Du, S.; Wang, H.; Xin, B.; Wang, J.; Shen, W.; Wei, W.; Guo, Z.; Shen, X. Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer. Oncotarget 2017, 8, 74406–74422.
- Chen, P.-C.; Tang, C.-H.; Lin, L.-W.; Tsai, C.-H.; Chu, C.-Y.; Lin, T.-H.; Huang, Y.-L. Thrombospondin-2 promotes prostate cancer bone metastasis by the up-regulation of matrix metalloproteinase-2 through down-regulating miR-376c expression. J. Hematol. Oncol. 2017, 10, 33.
- Du, R.; Tang, G.; Tang, Z.; Kuang, Y. Ectopic expression of CC chemokine receptor 7 promotes prostate cancer cells metastasis via Notch1 signaling. J. Cell. Biochem. 2019, 120, 9639–9647.
- Swenson-Fields, K.I.; Sandquist, J.C.; Rossol-Allison, J.; Blat, I.C.; Wennerberg, K.; Burridge, K.; Means, A.R. MLK3 limits activated Galphaq signaling to Rho by binding to p63RhoGEF. Mol. Cell 2008, 32, 43–56.
- Chen, J.; Gallo, K.A. MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res. 2012, 72, 4130–4140.
- Tsubouchi, A.; Sakakura, J.; Yagi, R.; Mazaki, Y.; Schaefer, E.; Yano, H.; Sabe, H. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell Biol. 2002, 159, 673–683.
- Swenson, K.I.; Winkler, K.E.; Means, A.R. A new identity for MLK3 as an NIMA-related, cell cycle-regulated kinase that is localized near centrosomes and influences microtubule organization. Mol. Biol. Cell 2003, 14, 156–172.
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284.
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017, 2925869.
- Rattanasinchai, C.; Gallo, K.A. MLK3 Signaling in Cancer Invasion. Cancers 2016, 8, 51.
More