Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Beatrix Zheng and Version 1 by Hassan Bousbaa.
The BUB3 protein plays a key role in the activation of the spindle assembly checkpoint (SAC), a ubiquitous surveillance mechanism that ensures the fidelity of chromosome segregation in mitosis and, consequently, prevents chromosome mis-segregation and aneuploidy. Besides its role in SAC signaling, BUB3 regulates chromosome attachment to the spindle microtubules. It is also involved in telomere replication and maintenance. Deficiency of the BUB3 gene has been closely linked to premature aging. Upregulation of the BUB3 gene has been found in a variety of human cancers and is associated with poor prognoses.
- BUB3
- spindle assembly checkpoint
- mitosis
- cancer
- senescence
- anticancer target
1. BUB3 in Aging
BUB3 shares extensive sequence homology with each of the four WD repeat motifs, and over the entire length of the RAE1 protein, indicative of functional similarity [31,38][1][2]. While BUB3 functions in the SAC pathway, RAE1 (also called Gle2 or mrnp41) is involved in mRNA export in interphase [38,39,40,41,42][2][3][4][5][6]. Binding to RAE1 is mediated by a GLEBS motif present in the nucleoporin Nup98 [41][5]. Strikingly, RAE1 also binds to the GLEBS motif of BUB1 [43][7]. The discovery that BUB3 also binds to the GLEBS motifs of the SAC proteins BUB1 and BUBR1 has led to the hypothesis that RAE1 might have a role as an SAC protein [43][7]. Homologous recombination-mediated mouse Rae1 gene disruption showed that the loss of a single Rae1 allele causes a SAC defect and chromosome mis-segregation. Besides the 34% identity and 52% similarity of the human RAE1 and BUB3, Bub3 haploinsufficient cells exhibit a strikingly similar mitotic phenotype, suggesting that RAE1 and BUB3 are functionally analogous, namely, by playing a specific or perhaps a redundant role in BUB1 targeting to unattached kinetochores and subsequent SAC activation [26,44][8][9]. Interestingly, double Rae1/Bub3 haploinsufficiency causes a much more severe chromosomal instability phenotype than single haploinsufficiencies, suggesting a cooperative role of RAE1 and BUB3 in regulating the SAC activities to prevent chromosomal mis-segregation [44][9]. Long-term phenotype analysis showed a reduced lifespan of mice harboring the combined Bub3 and Rae1 haploinsufficiency, with phenotypes associated with aging appearing early in double haploinsufficient mice, while mice with single Bub3 or Rae1 haploinsufficiency were viable and had a normal appearance [9,10,44][9][10][11]. Aneuploidy in single haploinsufficient Bub3 or Rae1 mice increased dramatically with age, and increased further in double Bub3/Rae1 haploinsufficient mice [10,44,45][9][11][12]. Curiously, mice with single or combined disruption of Bub3 and Rae1 were not predisposed to spontaneous tumorigenesis. Instead, Bub3/Rae1 haploinsufficiency caused early onset of cellular senescence, which was due to SAC weakening, rather than to aneuploidy itself. Since the age-associated phenotypes exhibited by haploinsufficient Bub3/Rae1 mice also occur in very old wild-type mice, then Rae1 and Bub3 were proposed to accelerate the aging process. Molecularly, haploinsufficient Bub3/Rae1 mice embryonic fibroblasts (MEFs) accumulate high levels of cellular senescence inductors, including p16, p19, p21, and p53, but, surprisingly, no major signs of apoptosis, suggesting that haploinsufficiency of Bub3 and Rae1 accelerates aging through induction of cellular senescence [9,10,44,45][9][10][11][12]. Significantly, and similarly to haploinsufficient Bub3/Rae1 mice, hypomorphic BubR1 mice develop several aging-associated phenotypes at a very young age, including cataracts, lordokyphosis, loss of subcutaneous fat, and impaired wound healing [46][13]. However, hypomorphic BubR1 mice had a much earlier onset of aging phenotypes, with many more senescent cells, than haploinsufficient Bub3/Rae1 mice, indicating that the rate of premature aging is correlated with the level of induction of senescence. Therefore, in addition to oncogenic transformation, accelerated aging seems to be another major biological manifestation of a weakened SAC [10,46][11][13]. What determines if it is oncogenic transformation or accelerated aging that will take place in a deficient SAC background is unknown. It might depend on the extent of SAC deficiency and/or SAC component depletion.
2. BUB3 in Cancer
Defects in SAC activity lead to chromosome mis-segregation, which is thought to be responsible, at least in part, for aneuploidy generation in human malignancies [1,47,48,49,50][14][15][16][17][18]. SAC deficiency is often associated with deregulated SAC genes [1,48,50][14][16][18]. WThe researchers examined the expression of BUB3 in various human cancer types. To this end, BUB3 gene expression and clinical data for 35 cancer types retrieved from the UALCAN data portal (http://ualcan.path.uab.edu/index.html, accessed on 24 December 2021) were analyzed [51][19]. BUB3 transcript levels were compared between cancers and normal tissue in 18 cancer types; 17 cancer types were excluded from the analysis due to lack of normal samples.
BUB3 protein levels are also elevated in a wide variety of human cancers compared to normal tissue (Figure 31). WThe researchers analyzed BUB3 protein levels in TP53-mutant cancers, as TP53-dependent SAC has been described [52,53,54][20][21][22]. TP53 is a transcription factor that acts as a tumor suppressor by inducing cell cycle arrest, cellular senescence, or apoptosis in response to cellular stresses, such as hypoxia, DNA and spindle damage [55][23]. TP53 gene mutations are universal across cancer types, and this contributes to human cancers in different ways [56][24]. The TP53 pathway regulates the expression of a network of genes that are targeted to respond to a variety of intrinsic and extrinsic stress signals to ensure, among other things, accurate DNA replication, chromosome segregation, and cell division [57][25]. Interestingly, in most of the cancer types analyzed, BUB3 levels are significantly higher in TP53-mutant cancers than in TP53-wild-type cancers, suggesting that wild-type TP53 represses BUB3 gene expression in physiological conditions, and that the TP53–BUB3 pathway may play an important role in carcinogenesis (Figure 31).
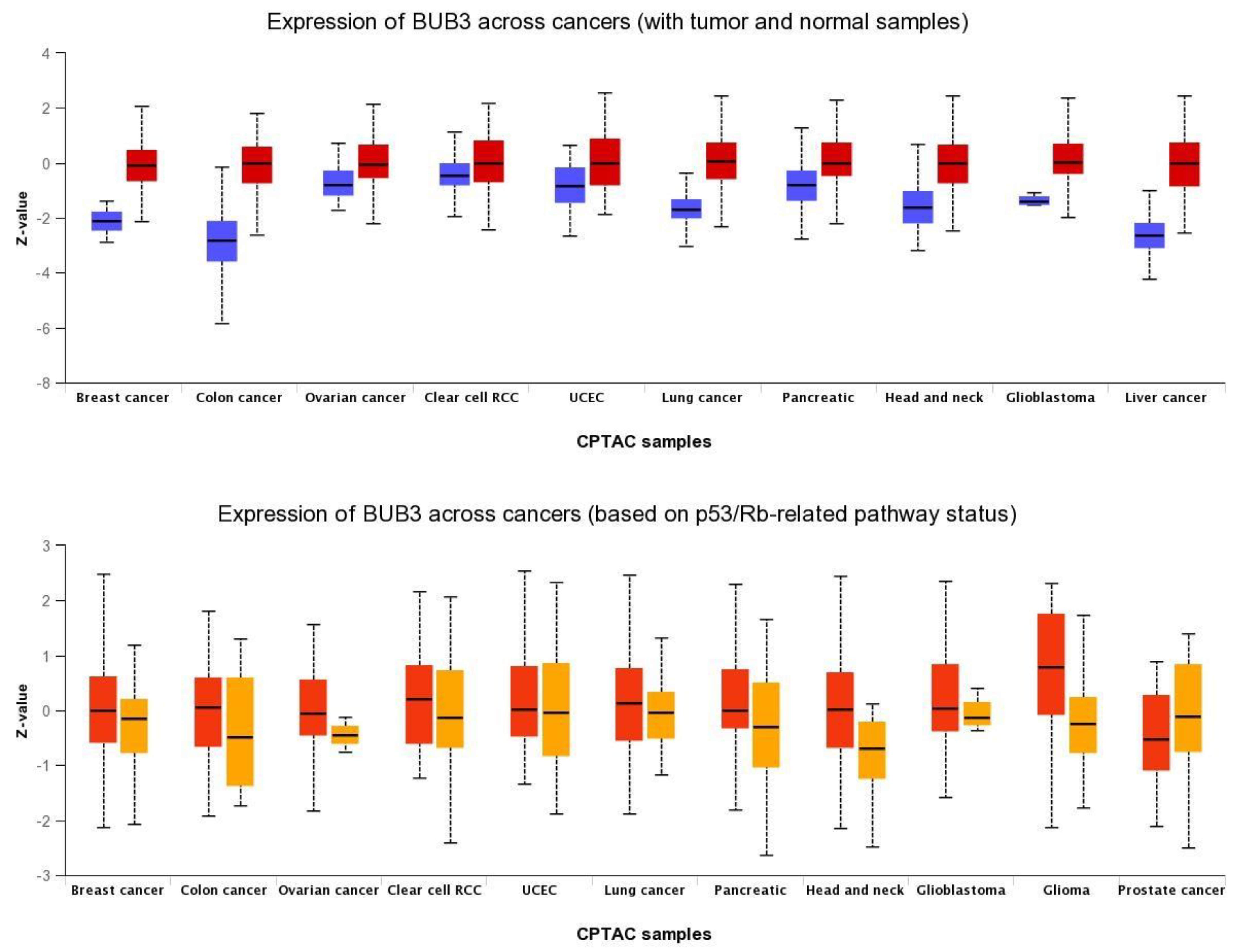
Figure 31. Pan-cancer view of expression of BUB3 protein across cancers. (Upper panel) Comparison between normal (blue) and primary tumors (red); (lower panel) Comparison between TP53-mutant (red) and TP53-non-mutant (orange) tumor samples. RCC: renal cell carcinoma; UCEC: Uterine corpus endometrial carcinoma; CPTAC: Clinical Proteomic Tumor Analysis Consortium. Data were retrieved from UALCAN portal (http://ualcan.path.uab.edu/index.html) on 24 December 2021.
Previous studies have reported BUB3 overexpression, at both RNA and protein levels, in a variety of human cancers compared with normal tissue. In most cancers, this upregulation was associated with poor prognoses. WThe researchers reported that BUB3 is upregulated and is associated with poor prognosis in oral squamous cell carcinoma [11][26]. The positive expression of cytoplasmic BUB3, together with that of cyclin B1 and the pituitary tumor-transforming gene 1, was significantly correlated with recurrence in prostate cancer [58][27]. BUB3 was upregulated in 79% of gastric cancers, being a proliferation-dependent phenomenon in gastric cancer [13][28]. BUB3 levels were reported to be higher in sarcoma samples, and higher expression levels of BUB3 were associated with lower overall and disease-free survival in patients with sarcomas [59][29]. High expression of BUB3 was associated with increased mortality in hepatocellular carcinoma [60][30]. In other studies, however, high protein expression of BUB3 in low-grade breast cancers was associated with longer overall survival, whereas lower expression resulted in poorer outcomes [61][31]. Upregulated BUB3 was also reported in breast cancer samples [62][32]. Polymorphism in the BUB3 gene was associated with the worst survival outcomes in early-stage non-small-cell lung cancer [63][33]. As with other SAC genes, epigenetic deregulation remains the most common alteration in the BUB3 gene, while mutations at the sequence levels are rather rare and confer no increased cancer risk [1][14]. For instance, genetic variation in the BUB3 gene did not affect familial breast cancer risk, and mutations in the BUB3 gene were shown to be rare in bladder tumors and glioblastomas [64,65,66][34][35][36]. Overall, these studies confirm that the overexpression of the BUB3 gene and protein is a common feature of human cancers, being associated with poor prognosis.
Why is BUB3 overexpressed in cancer cells? This question still remains unanswered. BUB3 and other SAC genes are frequently overexpressed in cancer, and such overexpression is correlated with chromosomal instability [67][37]. It was reported that loss of major tumor suppressor pathways, such as RB and TP53 pathways, can lead to transcriptional upregulation of SAC genes through E2F promoters and, subsequently, to chromosome mis-segregation [68,69,70][38][39][40]. As suggested by outhe researchers' analysis (Figure 31), TP53 loss could also lead to BUB3 upregulation, which should fuel chromosomal instability in cancer cells.
The role of BUB3 in carcinogenesis is still unclear. Contradictory results have been reported from animal models. For instance, haploinsufficiency of Bub3 causes an increase in chromosome instability in mice, but is not clearly associated with the frequency or the rate at which tumors appear in the animal [71][41]. Analysis of mice with reduced levels of Bub3 has shown that mice have significant increases in the number of aneuploid fibroblasts, and are predisposed to chemical-induced lung tumorigenesis rather than spontaneous tumor development [44][9]. A tumor suppressor role has been suggested for Bub3 in a Drosophila melanogaster tumorigenesis model derived from knocking down SAC genes [72][42]. Indeed, when transplanted into adult flies, Bub3-deficient tumors displayed neoplastic growth, widespread chromosomal aneuploidy, and high proliferative potential. Overall, these studies reveal that aneuploidy induced by BUB3 downregulation might not be sufficient to initiate tumorigenesis but might still facilitate it.
3. BUB3 as an Anticancer Therapeutic Target
For many years, the role of BUB3 has been reduced to the recruitment of its partners BUB1 and BUBR1 to unattached kinetochores. Probably for this reason, BUB3 has not been regarded as a potential anticancer target. Nevertheless, and as referred to above, BUB3 itself has a specific role in regulating kinetochore–microtubule attachments, and is involved in telomere replication maintenance and premature aging [5,8,10][11][43][44]. Importantly, the BUB3 gene is upregulated in most cancers studied, which is generally associated with poor outcomes. Thus, BUB3 is not just a simple partner, and its targeting deserves attention. Today, there are no small molecules against BUB3, and the unique attempt to target BUB3 makes use of RNAi [11][26]. InThe this study, wresearchers here have shown that RNAi-mediated inhibition of BUB3 was cytotoxic to OSCC cells and enhanced their chemosensitivity to cisplatin [11][26]. This antiproliferative activity of BUB3 inhibition against OSCC cells was recently confirmed by another group [73][45]. Very recently, wthe shresearchers showed that inhibition of BUB3 compromises glioblastoma cell proliferation, mainly through senescence induction rather than by apoptosis, suggesting that premature senescence can be a viable approach to restrain cancer propagation [74][46]. Thus, oligonucleotide-based targeting of BUB3 could be a viable therapeutic approach. However, small-molecule inhibitors should be a better option due to RNAi security and stability issues. As BUB3 is a non-enzyme protein, and, thus, an “undruggable target”, the development of an anti-BUB3 drug may be a challenging task. To circumvent this, one should design small molecules that target protein–protein interactions to interfere with biological processes by modulating the formation of protein–protein complexes. In this sense, targeting the interaction of BUB3 with BUB1 and BUBR1 is an attractive option. This would prevent MCC formation, leading to SAC inactivation, which is expected to kill cancer cells as a consequence of massive chromosome mis-segregation. Strategies to mimic Bub3/Rae1 haploinsufficiency in order to induce premature senescence of cancer cells should be explored. Indeed, cellular senescence has also been considered a suppressive mechanism of tumorigenesis, making therapy-induced senescence a plausible approach for cancer treatment, by irreversibly arresting the cell cycle [75][47].
References
- Taylor, S.S.; Ha, E.; McKeon, F. The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J. Cell Biol. 1998, 142, 1–11.
- Martinez-Exposito, M.J.; Kaplan, K.B.; Copeland, J.; Sorger, P.K. Retention of the BUB3 checkpoint protein on lagging chromosomes. Proc. Natl. Acad. Sci. USA 1999, 96, 8493–8498.
- Brown, J.A.; Bharathi, A.; Ghosh, A.; Whalen, W.; Fitzgerald, E.; Dhar, R. A mutation in the Schizosaccharomyces pombe rae1 gene causes defects in poly(A)+ RNA export and in the cytoskeleton. J. Biol. Chem. 1995, 270, 7411–7419.
- Murphy, R.; Watkins, J.L.; Wente, S.R. GLE2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol. Biol. Cell 1996, 7, 1921–1937.
- Pritchard, C.E.; Fornerod, M.; Kasper, L.H.; van Deursen, J.M. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J. Cell Biol. 1999, 145, 237–254.
- Bailer, S.M.; Siniossoglou, S.; Podtelejnikov, A.; Hellwig, A.; Mann, M.; Hurt, E. Nup116p and nup100p are interchangeable through a conserved motif which constitutes a docking site for the mRNA transport factor gle2p. EMBO J. 1998, 17, 1107–1119.
- Wang, X.; Babu, J.R.; Harden, J.M.; Jablonski, S.A.; Gazi, M.H.; Lingle, W.L.; de Groen, P.C.; Yen, T.J.; van Deursen, J.M. The mitotic checkpoint protein hBUB3 and the mRNA export factor hRAE1 interact with GLE2p-binding sequence (GLEBS)-containing proteins. J. Biol. Chem. 2001, 276, 26559–26567.
- Larsen, N.A.; Harrison, S.C. Crystal structure of the spindle assembly checkpoint protein Bub3. J. Mol. Biol. 2004, 344, 885–892.
- Babu, J.R.; Jeganathan, K.B.; Baker, D.J.; Wu, X.; Kang-Decker, N.; van Deursen, J.M. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 2003, 160, 341–353.
- Dai, W.; Wang, X. Aging in check. Sci. Aging Knowl. Environ. 2006, 2006, pe9.
- Baker, D.J.; Jeganathan, K.B.; Malureanu, L.; Perez-Terzic, C.; Terzic, A.; van Deursen, J.M.A. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J. Cell Biol. 2006, 172, 529–540.
- Kalitsis, P.; Earle, E.; Fowler, K.J.; Choo, K.H. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 2000, 14, 2277–2282.
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749.
- Silva, P.; Barbosa, J.; Nascimento, A.V.; Faria, J.; Reis, R.; Bousbaa, H. Monitoring the fidelity of mitotic chromosome segregation by the spindle assembly checkpoint. Cell Prolif. 2011, 44, 391–400.
- Marques, S.; Fonseca, J.; Silva, P.M.A.; Bousbaa, H. Targeting the spindle assembly checkpoint for breast cancer treatment. Curr. Cancer Drug Targets 2015, 15, 272–281.
- Diogo, V.; Teixeira, J.; Silva, P.M.A.; Bousbaa, H. Spindle Assembly Checkpoint as a Potential Target in Colorectal Cancer: Current Status and Future Perspectives. Clin. Colorectal Cancer 2017, 16, 1–8.
- Danielsen, H.E.; Pradhan, M.; Novelli, M. Revisiting tumour aneuploidy—The place of ploidy assessment in the molecular era. Nat. Rev. Clin. Oncol. 2016, 13, 291–304.
- Kops, G.J.P.L.; Weaver, B.A.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785.
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658.
- Chun, A.C.S.; Jin, D.-Y. Transcriptional regulation of mitotic checkpoint gene MAD1 by p53. J. Biol. Chem. 2003, 278, 37439–37450.
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305.
- Schvartzman, J.-M.; Duijf, P.H.G.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714.
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104.
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310.
- Silva, P.M.A.; Delgado, M.L.; Ribeiro, N.; Florindo, C.; Tavares, Á.A.; Ribeiro, D.; Lopes, C.; do Amaral, B.; Bousbaa, H.; Monteiro, L.S. Spindly and Bub3 expression in oral cancer: Prognostic and therapeutic implications. Oral Dis. 2019, 25, 1291–1301.
- Ersvær, E.; Kildal, W.; Vlatkovic, L.; Cyll, K.; Pradhan, M.; Kleppe, A.; Hveem, T.S.; Askautrud, H.A.; Novelli, M.; Wæhre, H.; et al. Prognostic value of mitotic checkpoint protein BUB3, cyclin B1, and pituitary tumor-transforming 1 expression in prostate cancer. Mod. Pathol. 2020, 33, 905–915.
- Grabsch, H.; Takeno, S.; Parsons, W.J.; Pomjanski, N.; Boecking, A.; Gabbert, H.E.; Mueller, W. Overexpression of the mitotic checkpoint genes BUB1, BUBR1, and BUB3 in gastric cancer—Association with tumour cell proliferation. J. Pathol. 2003, 200, 16–22.
- Long, Z.; Wu, T.; Tian, Q.; Carlson, L.A.; Wang, W.; Wu, G. Expression and prognosis analyses of BUB1, BUB1B and BUB3 in human sarcoma. Aging 2021, 13, 12395–12409.
- Liping, X.; Jia, L.; Qi, C.; Liang, Y.; Dongen, L.; Jianshuai, J. Cell Cycle Genes Are Potential Diagnostic and Prognostic Biomarkers in Hepatocellular Carcinoma. BioMed Res. Int. 2020, 2020, 6206157.
- Mukherjee, A.; Joseph, C.; Craze, M.; Chrysanthou, E.; Ellis, I.O. The role of BUB and CDC proteins in low-grade breast cancers. Lancet 2015, 385, S72.
- Yuan, B.; Xu, Y.; Woo, J.-H.; Wang, Y.; Bae, Y.K.; Yoon, D.-S.; Wersto, R.P.; Tully, E.; Wilsbach, K.; Gabrielson, E. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin. Cancer Res. 2006, 12, 405–410.
- Kang, H.G.; Yoo, S.S.; Choi, J.E.; Hong, M.J.; Do, S.K.; Jin, C.C.; Kim, S.; Lee, W.K.; Choi, S.H.; Lee, S.Y.; et al. Polymorphisms in mitotic checkpoint-related genes can influence survival outcomes of early-stage non-small cell lung cancer. Oncotarget 2017, 8, 61777–61785.
- Vaclavicek, A.; Bermejo, J.L.; Wappenschmidt, B.; Meindl, A.; Sutter, C.; Schmutzler, R.K.; Kiechle, M.; Bugert, P.; Burwinkel, B.; Bartram, C.R.; et al. Genetic variation in the major mitotic checkpoint genes does not affect familial breast cancer risk. Breast Cancer Res. Treat. 2007, 106, 205–213.
- Hernando, E.; Orlow, I.; Liberal, V.; Nohales, G.; Benezra, R.; Cordon-Cardo, C. Molecular analyses of the mitotic checkpoint components hsMAD2, hBUB1 and hBUB3 in human cancer. Int. J. Cancer 2001, 95, 223–227.
- Reis, R.M.; Nakamura, M.; Masuoka, J.; Watanabe, T.; Colella, S.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Mutation analysis of hBUB1, hBUBR1 and hBUB3 genes in glioblastomas. Acta Neuropathol. 2001, 101, 297–304.
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048.
- Schvartzman, J.-M.; Sotillo, R.; Benezra, R. Mitotic chromosomal instability and cancer: Mouse modelling of the human disease. Nat. Rev. Cancer 2010, 10, 102–115.
- Hernando, E.; Nahlé, Z.; Juan, G.; Diaz-Rodriguez, E.; Alaminos, M.; Hemann, M.; Michel, L.; Mittal, V.; Gerald, W.; Benezra, R.; et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 2004, 430, 797–802.
- Kabeche, L.; Compton, D.A. Checkpoint-Independent Stabilization of Kinetochore-Microtubule Attachments by Mad2 in Human Cells. Curr. Biol. 2012, 22, 638–644.
- Kalitsis, P.; Fowler, K.J.; Griffiths, B.; Earle, E.; Chow, C.W.; Jamsen, K.; Choo, K.H.A. Increased chromosome instability but not cancer predisposition in haploinsufficientBub3 mice. Genes Chromosom. Cancer 2005, 44, 29–36.
- da Silva, S.M.; Moutinho-Santos, T.; Sunkel, C.E. A tumor suppressor role of the Bub3 spindle checkpoint protein after apoptosis inhibition. J. Cell Biol. 2013, 201, 385–393.
- Logarinho, E.; Resende, T.; Torres, C.; Bousbaa, H. The human spindle assembly checkpoint protein Bub3 is required for the establishment of efficient kinetochore-microtubule attachments. Mol. Biol. Cell 2008, 19, 1798–1813.
- Li, F.; Kim, H.; Ji, Z.; Zhang, T.; Chen, B.; Ge, Y.; Hu, Y.; Feng, X.; Han, X.; Xu, H.; et al. The BUB3-BUB1 Complex Promotes Telomere DNA Replication. Mol. Cell 2018, 70, 395–407.e4.
- Zheng, J.; Li, T.-K.; Bao, Y.; Zhang, S.-X. Expression and clinical significance of Spindly and Bub3 in oral squamous cell carcinoma. Shanghai Kou Qiang Yi Xue 2020, 29, 528–532.
- Silva, P.M.A.; Nascimento, V.A.; Maritinho, O.; Reis, R.M.; Bousbaa, H. Targeting BUB3 in combination with paclitaxel inhibits proliferation of glioblastoma cells by enhancing cellular senescence. Sci. Lett. 2022, 1, 1.
- Lee, S.; Lee, J.-S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41.
More