Brain injury is a common cause of death and disability for people of all ages worldwide. Depending on the biomechanics, brain lesions may occur both in areas of the brain directly adjacent to the place of force application and in remote areas [4]. The mechanisms of hippocampal damage are of particular importance, since they underlie late complications of traumatic brain injury (TBI), such as epilepsy, depression and cognitive impairment. The mechanisms of reorganization of neuronal networks in the hippocampus include long-lasting chronic neuroinflammation and secondary damage to the nervous tissue. Responses and disturbances of the hypothalamic–pituitary–adrenal (HPA) axis may play a critical role in late post-traumatic pathology, in particular by modulation of synaptic activity and neuroinflammation in the hippocampus.
- brain trauma
- glucocorticoids
- corticosterone
- cortisol
- stress
- neuroinflammation
1. TBI, Its Late Consequences and the Hippocampus
2. HPA Axis in Patients with TBI
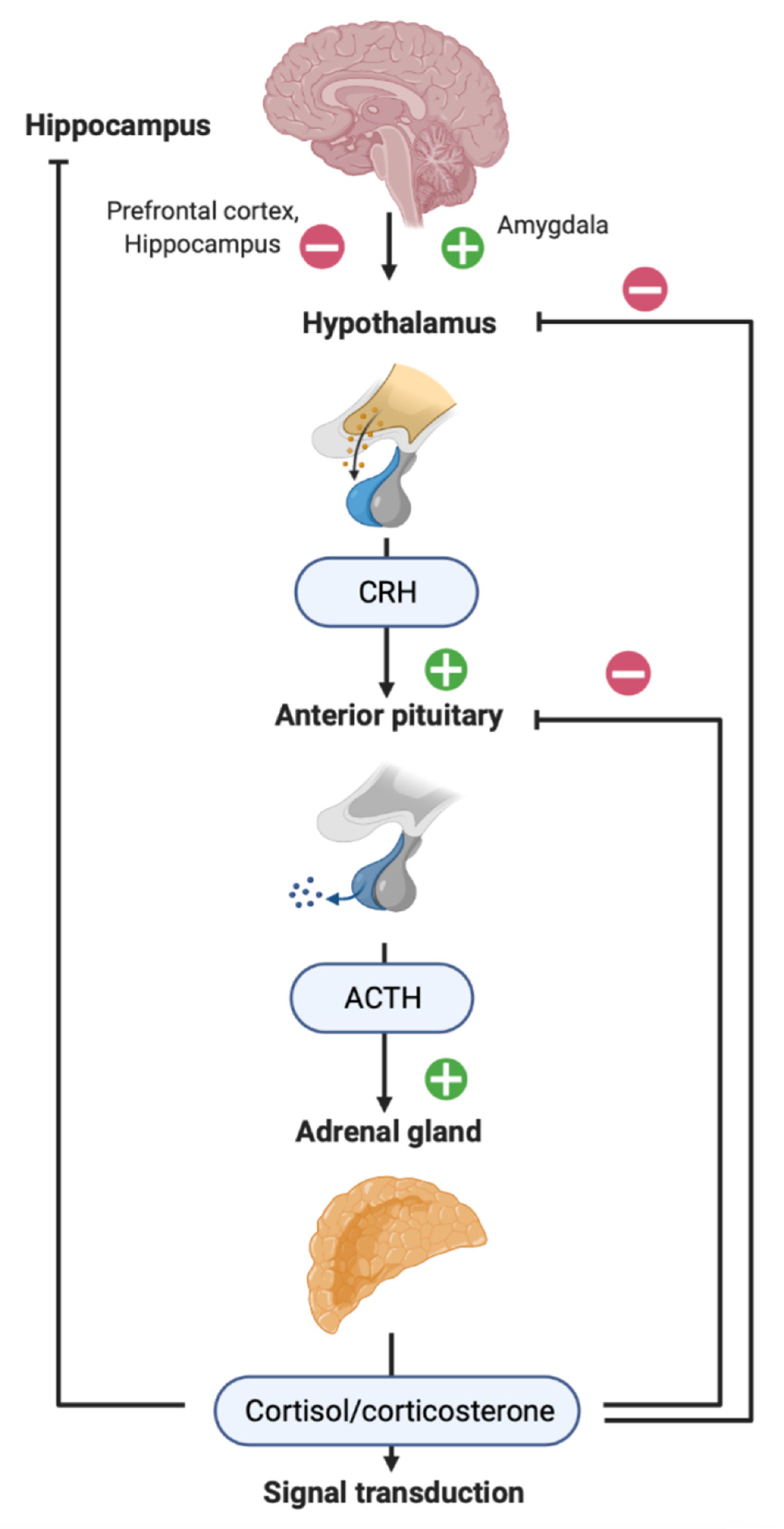
3. Neuroinflammation and TBI
Both primary and secondary mechanisms of brain damage discussed above are associated with neuroinflammatory response. Neuroinflammation is one of the essential mechanisms of brain damage modulated by GCs. Under normal conditions, microglial activity and cytokine release are intimately associated with neuroplasticity and memory; however, uncontrolled excessive neuroinflammation leads to cell death and further progression of brain pathology [99][28]. In pathological conditions, microglia play a crucial role in the expression of both pro-inflammatory (IL-1ß, IL-6, TNFα) and anti-inflammatory (IL-4, IL-10) cytokines, chemokines, as well as molecular fragments associated with damage (DAMP, damage-associated molecular patterns, such as HMGB1, ATP, S100ß). These substances enable microglial modulation of cyclooxygenase-2 and the components of the complement system [49][29]. Cytokines produced by inflammatory cells are released within minutes after TBI and alter the functioning of glutamate and GABAergic receptors, as well as potential-dependent ion channels, inhibit the reuptake of glutamate by astrocytes and provoke an increase in extracellular K+. Thus, cytokines can participate in hypersynchronization of neurons and the occurrence of epileptiform activity [100][30], as well as contribute to further neurodegeneration [101][31]. Microglial activation also occurs in remote areas of the brain, microglial properties and cytokine profile changing over time [102][32]. In the area of TBI, neutrophils and other cells of the immune system are recruited as well. These cells, along with glia, take part in the production of cytokines, chemokines, free radicals, prostaglandins and components of the complement system. The profile of peripheral immune system cells changes over time. Neutrophils first appear in the focus of injury; after 3–5 days they are replaced by mononuclear leukocytes and, to a lesser degree, by T cells, dendritic cells and natural killers [49][29]. The peripheral immune system is also activated. It has been shown that 1 day after TBI, the number of CD4+ and CD8+ T cells in rat spleen increases, indicating an activation of adaptive immunity. Suppression of adaptive immunity improves TBI outcomes [103][33]. Thus, autoimmune mechanisms are involved in the development of post-traumatic pathology [49][29], though their role has not been studied in detail yet. Astrocytic gliosis in the neocortex develops about 1 week after TBI and, in the long-term period of injury astrogliosis, serves as an important histopathological marker of hippocampal sclerosis [104,105,106][34][35][36]. Astrocyte dysfunction may be involved in increasing the excitability of neurons and circuit reorganization via several mechanisms. Astrocytes normally participate in the utilization of extracellular K+ (due to active transport into the cell and distribution through the astrocyte system) and utilization/metabolism of glutamate. Changes in K+ homeostasis and an increase in its concentration lead to a decrease in neuronal excitability threshold, while impairment of glutamate utilization results in an increase in its toxic effects. In addition, astrocytes play an important role in water homeostasis of the brain [107][37] and form the brain glymphatic system involved in the development and resorption of edema, transport of metabolites and immune cells [108][38]. The transition from acute activation of the brain immune system to chronic neuroinflammation in TBI is the subject of quite a few studies [5,17,109][12][39][40]. Chronic neuroinflammation caused by TBI induces progressive edema and neurodegeneration associated with cognitive and emotional disorders [110][41]. The first week after TBI is an important time interval, day 7 being considered a borderline between acute and chronic post-traumatic changes. It is noteworthy that edema resorption and the early development of astrogliosis in the focus of direct impact to the neocortex was shown 7 days after TBI [104,105,106][34][35][36].4. Neuroinflammation and GCs
Chronic neuroinflammation is a recognized consequence of chronic stress; its definitive association with GCs is rigorously discussed but still remains obscure [111][42]. The available data indicate dual effects of GCs, both anti- and pro-inflammatory. Suppression of inflammation is among the well-established systemic effects of GCs. This ability of GCs is widely used in clinical practice for treatment of inflammatory and autoimmune diseases. The activation of GRs and MRs in peripheral tissues results in inhibition of immune cell activity and induction of apoptosis in lymphocytes [112][43]. GCs also inhibit inflammation via several other mechanisms, including inhibition of tissue infiltration by cells from the blood, inhibition of cytokine expression, changes of lymphocyte functioning and others [111][42]. In the brain, GCs realize either pro- or anti-inflammatory properties depending on the degree and duration of exposure, external factors preceding injury, injury characteristics and the specific brain region [31,70,111][15][42][44]. The order and time period between GC increase and immune challenge may be important for the effects of GCs on neuroinflammation (Figure 2). This was confirmed in a study with administration of GCs and lipopolysaccharide (LPS, immunogenic component of Gram-negative bacteria) in a different order [113][45]. If GCs were injected prior to LPS (2 and 24 h), they potentiated pro-neuroinflammatory effects (TNFa, IL-1b, IL-6 expression). In contrast, GCs injected 1 h after LPS had an anti-inflammatory action in the brain. LPS injection directly into the hippocampus of the stressed animals also increased the number of reactive microglial cells and expression of pro-inflammatory cytokines [114][46] as compared to non-stressed animals. The second factor affecting GCs action is the duration of their exposure (Figure 2). Many groups have demonstrated that chronic stress is definitely a pro-inflammatory condition [111][42]. Chronic stress potentiated LPS-induced activation of several pro-inflammatory pathways, including nuclear factor kappa B (NF-κB) [115][47], and increased basal activation of other intracellular pathways, such as ERK1/2, p38, SAPK/JNK and AKT [116][48].

References
- Annegers, J.F.; Hauser, W.A.; Coan, S.P.; Rocca, W.A. A population-based study of seizures after traumatic brain injuries. N. Engl. J. Med. 1998, 338, 20–24.
- Gupta, P.K.; Sayed, N.; Ding, K.; Agostini, M.A.; Van Ness, P.C.; Yablon, S.; Madden, C.; Mickey, B.; D’Ambrosio, R.; Diaz-Arrastia, R. Subtypes of Post-Traumatic Epilepsy: Clinical, Electrophysiological, and Imaging Features. J. Neurotrauma 2014, 31, 1439–1443.
- Malmgren, K.; Thom, M. Hippocampal sclerosis-Origins and imaging. Epilepsia 2012, 53, 19–33.
- Englander, J.; Bushnik, T.; Duong, T.T.; Cifu, D.X.; Zafonte, R.; Wright, J.; Hughes, R.; Bergman, W. Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Arch. Phys. Med. Rehabil. 2003, 84, 365–373.
- Haltiner, A.M.; Temkin, N.R.; Dikmen, S.S. Risk of seizure recurrence after the first late posttraumatic seizure. Arch. Phys. Med. Rehabil. 1997, 78, 835–840.
- Temkin, N.R. Risk Factors for Posttraumatic Seizures in Adults. Epilepsia 2003, 44, 18–20.
- Pohlmann-Eden, B.; Bruckmeir, J. Predictors and dynamics of posttraumatic epilepsy. Acta Neurol. Scand. 1997, 95, 257–262.
- Bombardier, C.H. Rates of Major Depressive Disorder and Clinical Outcomes Following Traumatic Brain Injury. JAMA 2010, 303, 1938.
- Gulyaeva, N.V. Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry 2019, 84, 1306–1328.
- Malykhin, N.V.; Carter, R.; Seres, P.; Coupland, N.J. Structural changes in the hippocampus in major depressive disorder: Contributions of disease and treatment. J. Psychiatry Neurosci. 2010, 35, 337–343.
- Hesdorffer, D.C.; Ishihara, L.; Mynepalli, L.; Webb, D.J.; Weil, J.; Hauser, W.A. Epilepsy, suicidality, and psychiatric disorders: A bidirectional association. Ann. Neurol. 2012, 72, 184–191.
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191.
- de Kloet, E.R.; Karst, H.; Joëls, M. Corticosteroid hormones in the central stress response: Quick-and-slow. Front. Neuroendocrinol. 2008, 29, 268–272.
- Maggio, N.; Segal, M. Corticosteroid Regulation of Synaptic Plasticity in the Hippocampus. Sci. World J. 2010, 10, 462–469.
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2019, 44, 1306–1322.
- Kusmenkov, T.; Braunstein, M.; Schneider, H.; Bidlingmaier, M.; Prall, W.; Flatz, W.; Boecker, W.; Bogner, V. Initial free cortisol dynamics following blunt multiple trauma and traumatic brain injury: A clinical study. J. Int. Med. Res. 2019, 47, 1185–1194.
- Kakati, A.; Devi, B.I.; Bhadrinarayan, V.; Kalra, P.; Shukla, D. Endocrine dysfunction following traumatic brain injury in acute stage. Indian J. Neurotrauma 2013, 10, 92–96.
- Rao, T.P. A study of serum cortisol levels in acute head injury patients. J. Basic Clin. Physiol. Pharmacol. 2020, 32, 20190136.
- Agha, A.; Rogers, B.; Mylotte, D.; Taleb, F.; Tormey, W.; Phillips, J.; Thompson, C.J. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin. Endocrinol. 2004, 60, 584–591.
- Bensalah, M.; Donaldson, M.; Aribi, Y.; Iabassen, M.; Cherfi, L.; Nebbal, M.; Medjaher, M.; Haffaf, E.; Abdennebi, B.; Guenane, K.; et al. Cortisol evaluation during the acute phase of traumatic brain injury-A prospective study. Clin. Endocrinol. 2018, 88, 627–636.
- Bernard, F.; Outtrim, J.; Lynch, A.G.; Menon, D.K.; Matta, B.F. Hemodynamic Steroid Responsiveness is Predictive of Neurological Outcome After Traumatic Brain Injury. Neurocrit. Care 2006, 5, 176–179.
- Tanriverdi, F.; Schneider, H.J.; Aimaretti, G.; Masel, B.E.; Casanueva, F.F.; Kelestimur, F. Pituitary Dysfunction After Traumatic Brain Injury: A Clinical and Pathophysiological Approach. Endocr. Rev. 2015, 36, 305–342.
- Saichan, X.; Wei, C.; Qinglong, F.; Jun, W.; Lei, X. Plasma cortisol as a noninvasive biomarker to assess severity and prognosis of patients with craniocerebral injury. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3835–3838.
- Sörbo, A.; Eiving, I.; Theodorsson, E.; Rydenhag, B.; Jonsdottir, I.H. Pre-traumatic conditions can influence cortisol levels before and after a brain injury. Acta Neurol. Scand. 2020, 141, 342–350.
- Spikman, J.M.; van der Horn, H.J.; Scheenen, M.E.; de Koning, M.E.; Savas, M.; Langerak, T.; van Rossum, E.F.C.; van der Naalt, J. Coping with stress before and after mild traumatic brain injury: A pilot hair cortisol study. Brain Inj. 2021, 35, 871–879.
- Bay, E.; Sikorskii, A.; Gao, F. Functional Status, Chronic Stress, and Cortisol Response After Mild-to-Moderate Traumatic Brain Injury. Biol. Res. Nurs. 2009, 10, 213–225.
- Bay, E.; Hagerty, B.; Williams, R.A.; Kirsch, N. Chronic Stress, Salivary Cortisol Response, Interpersonal Relatedness, and Depression Among Community-Dwelling Survivors of Traumatic Brain Injury. J. Neurosci. Nurs. 2005, 37, 4–14.
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153.
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting traumatic brain injury: From molecular mechanisms to therapeutic interventions. Biomedicines 2020, 8, 389.
- Vezzani, A.; Auvin, S.; Ravizza, T.; Aronica, E. Glia-Neuronal Interactions in Ictogenesis and Epileptogenesis: Role of Inflammatory Mediators. In Jasper’s Basic Mechanisms of the Epilepsies , 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M., Olsen, R., Delgado-Escueta, A., Eds.; Oxford University Press: Oxford, UK, 2012; ISBN 9780199746545.
- Vezzani, A.; Balosso, S.; Ravizza, T. Inflammation and epilepsy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 107, pp. 163–175. ISBN 9780444528988.
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208.
- Tobin, R.P.; Mukherjee, S.; Kain, J.M.; Rogers, S.K.; Henderson, S.K.; Motal, H.L.; Rogers, M.K.N.; Shapiro, L.A. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol. Commun. 2014, 2, 143.
- Hicks, R.; Soares, H.; Smith, D.; McIntosh, T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996, 91, 236–246.
- Cortez, S.C.; McIntosh, T.K.; Noble, L.J. Experimental fluid percussion brain injury: Vascular disruption and neuronal and glial alterations. Brain Res. 1989, 482, 271–282.
- Lescot, T.; Fulla-Oller, L.; Po, C.; Chen, X.R.; Puybasset, L.; Gillet, B.; Plotkine, M.; Meric, P.; Marchand-Leroux, C. Temporal and Regional Changes after Focal Traumatic Brain Injury. J. Neurotrauma 2010, 27, 85–94.
- Xu, S.; Sun, Q.; Fan, J.; Jiang, Y.; Yang, W.; Cui, Y.; Yu, Z.; Jiang, H.; Li, B. Role of Astrocytes in Post-traumatic Epilepsy. Front. Neurol. 2019, 10, 1149.
- Thrane, A.S.; Rangroo Thrane, V.; Nedergaard, M. Drowning stars: Reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014, 37, 620–628.
- Tapp, Z.M.; Godbout, J.P.; Kokiko-Cochran, O.N. A Tilted Axis: Maladaptive Inflammation and HPA Axis Dysfunction Contribute to Consequences of TBI. Front. Neurol. 2019, 10, 345.
- D’amico, R.; Salinaro, A.T.; Fusco, R.; Cordaro, M.; Impellizzeri, D.; Scuto, M.; Ontario, M.L.; Dico, G.L.; Cuzzocrea, S.; Di Paola, R.; et al. Hericium erinaceus and coriolus versicolor modulate molecular and biochemical changes after traumatic brain injury. Antioxidants 2021, 10, 898.
- Chiu, C.-C.; Liao, Y.-E.; Yang, L.-Y.; Wang, J.-Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.-Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49.
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry 2021, 86, 156–167.
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247.
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009, 64, 33–39.
- Frank, M.G.; Miguel, Z.D.; Watkins, L.R.; Maier, S.F. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain. Behav. Immun. 2010, 24, 19–30.
- Espinosa-Oliva, A.M.; de Pablos, R.M.; Villarán, R.F.; Argüelles, S.; Venero, J.L.; Machado, A.; Cano, J. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol. Aging 2011, 32, 85–102.
- Munhoz, C.D. Chronic Unpredictable Stress Exacerbates Lipopolysaccharide-Induced Activation of Nuclear Factor- B in the Frontal Cortex and Hippocampus via Glucocorticoid Secretion. J. Neurosci. 2006, 26, 3813–3820.
- Munhoz, C.D.; Sorrells, S.F.; Caso, J.R.; Scavone, C.; Sapolsky, R.M. Glucocorticoids Exacerbate Lipopolysaccharide-Induced Signaling in the Frontal Cortex and Hippocampus in a Dose-Dependent Manner. J. Neurosci. 2010, 30, 13690–13698.
- Komoltsev, I.G.; Tret’yakova, L.V.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Butuzov, A.V.; Novikova, M.R.; Kvichansky, A.A.; Moiseeva, Y.V.; Onufriev, M.V.; et al. Neuroinflammatory Cytokine Response, Neuronal Death, and Microglial Proliferation in the Hippocampus of Rats During the Early Period After Lateral Fluid Percussion-Induced Traumatic Injury of the Neocortex. Mol. Neurobiol. 2021, 59, 1151–1167.
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Onufriev, M.V.; Moiseeva, J.V.; Novikova, M.R.; Gulyaeva, N.V. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. Int. J. Mol. Sci. 2021, 22, 5883.
- Tretyakova, L.V.; Kvichansky, A.A.; Bolshakov, A.P.; Gulyaeva, N.V. Dexamethasone Modulates Lipopolysaccharide-Induced Expression of Proinflammatory Cytokines in Rat Hippocampus. Neurochem. J. 2021, 15, 302–307.
- Skupio, U.; Tertil, M.; Sikora, M.; Golda, S.; Wawrzczak-Bargiela, A.; Przewlocki, R. Behavioral and molecular alterations in mice resulting from chronic treatment with dexamethasone: Relevance to depression. Neuroscience 2015, 286, 141–150.
- Frank, M.G.; Hershman, S.A.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 2014, 40, 191–200.
- Fenn, A.M.; Gensel, J.C.; Huang, Y.; Popovich, P.G.; Lifshitz, J.; Godbout, J.P. Immune Activation Promotes Depression 1 Month After Diffuse Brain Injury: A Role for Primed Microglia. Biol. Psychiatry 2014, 76, 575–584.
- Dong, T.; Zhi, L.; Bhayana, B.; Wu, M.X. Cortisol-induced immune suppression by a blockade of lymphocyte egress in traumatic brain injury. J. Neuroinflamm. 2016, 13, 197.
- Raefsky, S.M.; Mattson, M.P. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic. Biol. Med. 2017, 102, 203–216.