Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Santiago Cuevas and Version 2 by Amina Yu.
The NLRP3 inflammasome is mainly expressed in myeloid cells, such as macrophages. However, it has also been described in other types of cells, such as endothelial cells, hepatocytes, or even hepatic stellate cells (HSCs); and its activation inducing pyroptosis has been associated with inflammation, fibrosis, and cell death in the liver.
- liver diseases
- NLRP3 inflammasome
- Nrf2
- ROS
1. Inflammasomes and Pyroptotic Cell Death
Inflammasomes are multi-protein signalling complexes that regulate the activation of inflammatory responses to microbial infection, and cellular damage, in addition to recognition of signals produced during altered homeostasis. An inflammasome is composed of an adaptor, an effector, and a sensor protein [1]. The assembly of inflammasomes is mediated by a sensor protein, which is usually a pattern recognition receptor (PRR) that oligomerizes following induction of activation by damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), or homeostasis-altering molecular processes (HAMPs). Inflammasome activation results in the proteolytic cleavage and activation of pro-inflammatory Caspase-1, which triggers pro-inflammatory cytokines and induces pyroptotic cell death [2].
Based on their structural domains and their capacity to assemble inflammasomes, there are different receptor proteins that have been confirmed to be the sensor proteins for these complexes. The most focstusdied are those formed by members of the nucleotide-binding oligomerization domain (NOD), leucine-rich repeat-containing receptor (NLR) family, comprising NLRP1, NLRP3, NLRP6, and NLRC4. These receptors have been documented as suitable for the formation of inflammasomes. Additionally, two non-NLR proteins, absent in melanoma 2 (AIM2) and Pyrin, also form functional inflammasomes [3]. NLRP3 is the most focstusdied inflammasome as its dysregulation has been associated with the pathophysiology of the auto-inflammatory cryopyrin-associated periodic syndrome (CAPS), the development of chronic inflammatory conditions, neurodegenerative disorders, and metabolic diseases, including liver pathologies and fibrosis [4].
The first step of the canonical NLRP3 inflammasome activation involves the stimulation of different receptors, including Toll-like receptors (TLRs), and activation of the nuclear factor kappa B (NF-kB). The activation of these molecules, on the one hand, increases the transcription of NLRP3, pro-IL1B, and pro-IL18; while on the other hand, it induces post-translational modifications of NLRP3 in the form of (de-)ubiquitination or (de-)phosphorylation [5][6][5,6], necessary for its activation (Figure 1). A subsequent signal is required to induce activation of the NLRP3 inflammasome multiprotein complex. This second signal is usually mediated by DAMPs, such as crystalline particles (e.g., uric acid or cholesterol crystals) or detection of extracellular adenosine triphosphate (ATP) by the purinergic P2X7 receptor [7]. This signal is related to lysosomal destabilization or mitochondrial dysfunction, which induces the formation of reactive oxygen species (ROS), cellular metabolic changes, potassium (K+) efflux, and calcium (Ca2+) influx, that are upstream mechanisms for NLRP3 activation in response to most triggers [8] (Figure 1). The downstream signalling induced by these DAMPs in primed macrophages involves the activation of NLRP3 oligomers that recruit the adaptor inflammasome protein called apoptosis-associated speck-like protein containing caspase activation and recruitment domain (ASC) (Figure 1). This adaptor protein consists of two death-fold domains: a pyrin domain (PYD) and caspase activation and recruitment domain (CARD). ASC is recruited to the NLRP3 activated oligomer via PYD-PYD homotypic domain interaction. Further PYD-PYD interactions result in the formation of ASC filaments. The effector zymogen pro-Caspase-1 is then recruited to these ASC filaments by CARD-CARD interaction, and the close proximity of pro-Caspase-1 zymogens thereby induces an auto-proteolytic activation of Caspase-1 within the inflammasome complex, initiating the formation of the catalytically active protease Caspase-1 (Figure 1). Caspase-1 is the effector protein of the inflammasome complex that cleaves the preforms of the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 into their mature and bioactive forms [9]. In addition, Caspase-1 cleaves Gasdermin D (GSDMD) and releases its necrotic and cytotoxic N-terminal domain from its repressor C-terminal domain [10][11][10,11] (Figure 1). Once GSDMD auto-inhibition is disrupted, the N-terminal domain of GSDMD binds to the plasma membrane and forms pores following homo-oligomerisation [12] (Figure 1). Both mature IL-1β and IL-18 are released through the GSDMD pore, as this pore has a negative conduit, and both cytokines present a basic surface after processing by Caspase-1 [13]. Upon permeabilization of the plasma membrane by GSDMD pores, cells undergo pyroptosis, a lytic process mediated by the oligomerization of the nerve injury-induced protein 1 [14]. During pyroptosis, other intracellular components, in addition to IL-1β and IL-18, are also released, including the alarmin high mobility group box 1 (HMGB1), mitochondrial DNA, or inflammasome oligomers [15][16][15,16], thereby resulting in a highly pro-inflammatory environment [17] (Figure 1). Various studies have associated excessive pyroptosis with multiple diseases, such as cardiovascular or immune diseases, disseminated intravascular coagulation in septic patients, and liver disorders [18][19][18,19].
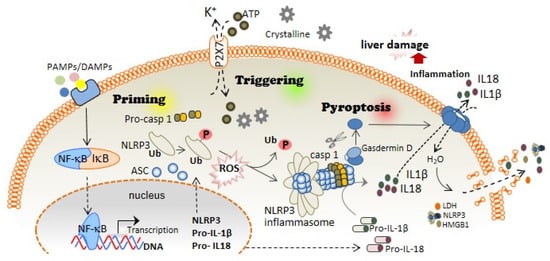
Figure 1. NLRP3 inflammasome and pyroptosis activation induces liver damage. The canonical activation of the NLRP3 inflammasome is differentiated in two steps: priming and triggering. Priming; PAMPs and DAMPs stimulate TLR and other receptors, which induce the NF-κB translocation to the cell nucleus, thus increasing the expression of the different inflammasome components: NLRP3, pro-IL-1β, and pro-IL-18 expression. During priming signaling, NLRP3 also undergoes different post-translational modifications that facilitate its activation, including phosphorylation/dephosphorylation and de-ubiquitination. Triggering; DAMPs such as extracellular ATP or crystalline structures induce the activation of the NLRP3 inflammasome oligomer, which leads to the activation of Caspase-1. Caspase-1 cleaves Gasdermin D in addition to pro-IL-1β and pro-IL-18, which turn into mature IL-1β and IL-18. Pyroptosis; The N-terminal fragment of Gasdermin D generated after Caspase-1 cleavage forms oligomeric pores in the plasma membrane allowing the release of pro-inflammatory cytokines such as IL-1β and IL-18 into the extracellular space. Additionally, Gasdermin D pores lead to water influx into the cell, cell swelling, and cell lysis mediated by the ninjury-1 protein, thus increasing the inflammatory response by releasing inflammatory products from the intracellular space.
The NLRP3 inflammasome is mainly expressed in myeloid cells, such as macrophages. However, it has also been described in other types of cells, such as endothelial cells, hepatocytes, or even hepatic stellate cells (HSCs); and its activation inducing pyroptosis has been associated with inflammation, fibrosis, and cell death in the liver [20].
2. Inflammasome and Pyroptosis as an Essential Inflammatory Pathway in Liver Diseases
Liver diseases are important contributors to global morbidity and mortality and are implicated in more than 45% of deaths in developed countries [21]. Diseases of the liver include conditions that cause any disturbance of its function resulting in illness. Sterile inflammation (in the absence of any pathogenic microorganisms) is a hallmark of the major diseases affecting the liver, including liver fibrosis, which results from chronic liver damage. The main causes of liver fibrosis in industrialized countries include alcohol abuse, non-alcoholic fatty liver disease (NAFLD), including non-alcoholic steatohepatitis (NASH), and also chronic viral infection. Liver fibrosis can lead to end-stage cirrhosis, where the liver parenchyma is substituted by scar tissue, wherein liver transplantation is the only option to recover normal liver functions [22]. In the presence of liver injury, some pro-inflammatory cytokines such as IL-1β are released, mainly by macrophages, activating HSCs through IL-1 receptor, turning into myofibroblasts and releasing a large amount of extracellular matrix, inducing the formation of scar tissue and leading to liver fibrosis [23]. Therefore, the NLRP3 inflammasome is considered to be a critical pathway for pro-inflammatory cytokine release in the liver and is strongly involved in the pathogenesis of liver fibrogenesis [24]. Constitutive activation of NLRP3 in an Nlrp3A350v mutant mouse down-regulates metabolic pathways in hepatocytes and shifts HSCs toward a profibrotic state with an expression of collagen and extracellular matrix regulatory genes [25].
NLRP3 inflammasome is present in the cytoplasm of HSCs and is activated by various agents such as PAMPs (S. mansoni, E. coli, S. japonicum) [26][27][28][26,27,28] or through receptors such as P2X7R [29], angiotensin II receptors [30] or growth factor receptors [31]. In addition, NLRP3 activation in primary human hepatocytes leads to pyroptotic cell death and the release of inflammasome oligomeric particles into the extracellular space, resulting in the activation of HSCs by particle internalization [32]. In contrast, treatment with IL-10 and Jinlida, a traditional Chinese medicine, decreases the protein levels of NLRP3, IL1β, IL-18, and caspase-1, thus inhibiting the hepatocyte pyroptosis that ameliorated liver dysfunction [33][34][33,34]. In this regard, the regulation of pyroptosis by stress-inducible proteins such as Sestrin2, and transcription factors such as Ikaros together with SIRT1, has been reported to protect against liver injury through the downregulation of the inflammasome proteins [35][36][35,36]. More specifically, ia recent wasstudy pointeds to how treatment with phenethyl isothiocyanate, present as a natural compound in cruciferous vegetables and related to anti-cancer properties [37], can alleviate liver injury by reducing hepatocyte pyroptosis via direct inhibition of the cysteine 191 (Cys191) of GSDMD, which is necessary for the formation of GSDMD pores [38][39][38,39]. All of these results highlight the key role of pyroptosis in liver disease after NLRP3 inflammasome activation.
Numerous onstudies have demonstrated a link between the NLRP3 inflammasome and NAFLD [40], the condition in which fat builds up in the liver. NLRP3 inflammasome activation was indispensable in the fibrotic response in a murine model of NASH. In this regard, a methionine- and choline-deficient diet or high-fat diet evoked the overexpression of the inflammasome components NLRP3, ASC, caspase-1, and IL-1β [41]. Moreover, Nlrp3 knockout mice fed to induce NASH showed a clear reduction in myeloid cell infiltration with the consequent decrease in liver inflammation and fibrosis [41]. Pyroptosis is involved in NASH development; that is, Gsdmd deficiency alleviates steatosis and inflammation, whereas Gsdmd overexpression promotes liver fibrosis [42].
Alcoholic liver disease (ALD), associated with long-term alcohol abuse, evolves from steatosis to severe stages such as alcoholic steatohepatitis (ASH) and alcoholic cirrhosis [43]. Similar to NAFLD, high levels of NLRP3 inflammasome and caspase-1 activation, and even serum IL-1β concentration, were found in ethanol-fed murine models [44]. The development of alcoholic steatosis has been associated with Kupffer cells (KCs), which present a significantly higher expression of inflammasome components compared to hepatocytes, and, in this regard, the silencing of the Casp1 gene in KCs resulted in protection against steatosis, similar to that found in global Casp1 knock-out mice [45][46][45,46]. Likewise, hepatocytes injured by chronic ethanol exposure induce the release of ATP and uric acid, which are classical DAMPs able to activate the NLRP3 inflammasome in KCs through the induction of mitochondrial ROS [47]. In this regard, alcoholic steatosis development has been prevented in P2x7r knock-out mice, in whom ATP signalling is blocked, or when uric acid is degraded by uricase treatment [45]. A mechanism of NLRP3 regulation of thioredoxin in response to ethanol has been discovered in ALD from human and mouse models. Both cases showed Nlrp3, Pycard (the ASC coding gene), and Il1b upregulation followed by high serum IL-1β levels compared to control animals [48]. These results are supported by the increased mRNA levels of NLRP3 inflammasome components, pro-inflammatory cytokines, and profibrotic genes in patients with alcohol-induced liver damage [49]. Moreover, prolonged alcohol intake is also associated with an altered gut microbiome, which is linked with intestinal dysbiosis, which in turn can lead to the translocation of bacterial products and bacteria from the gut to the liver, thus evoking a favourable environment for the activation of NLRP3 inflammasome [50][51][50,51]. Likewise, inhibition of poly (ADP-ribose) polymerase-1, a key mediator of liver inflammation and fibrosis [52], which protects against both NASH and ASH [53], can be directly related to the modulation of NLRP3 inflammasome and pyroptosis activation [54]. Furthermore, GSDMD plays a significant role in the evolution of liver steatosis into ASH. An increase in GSDMD was observed in the livers of animals with ASH compared to steatotic or control animals [55].
The hepatitis B (HBV) and C (HCV) viruses are associated with cirrhosis and hepatocellular carcinoma, where liver inflammation is observed in different stages of the disease [56]. In HBV, where the hepatitis X protein has a key role in liver inflammation by inducing the release of ROS, the NLRP3 inflammasome is activated and thus increases the expression of IL1B, CASP1, and NLRP3 genes in the liver [57], followed by the secretion of ASC, IL-1β, IL-18, and HMGB1 in an environment of oxidative stress in hepatocytes [58]. This result is supported by high levels of IL-1β and IL-18 in plasma from patients with cirrhosis resulting from HBV [59]. Moreover, it has been shown that the HBV e antigen helps the virus to escape the host immune response by inhibiting NLRP3 inflammasome activation [60], while the hepatitis B core antigen seems to have a pro-inflammatory role by upregulating the expression and activation of the NLRP3 inflammasome, suggesting that the inflammasome is primed and activated in the first phases of infection [61]. Similarly, the HCV core protein has been shown to elicit NLRP3 inflammasome activation after the release of TNF-α with the consequent release of IL-1β by KCs to boost the inflammatory response of the liver [62][63][62,63].
In summary, inflammation is a major component in the progression of liver diseases, with important involvement of the NLRP3 inflammasome and pyroptosis. Therefore, the therapeutic targeting of NLRP3 or the proteins that accompany the formation of the NLRP3 inflammasome (such as ASC or Caspase-1) or even the released pro-inflammatory cytokines and the pore-forming protein GSDMD has garnered increasing consideration from the experts in the field and is an expanding area of research.
3. The Association of Oxidative Stress with NLRP3 Inflammasome Activation and Liver Diseases
Oxidative stress is characterised by the production of reactive oxygen and nitrogen species (ROS and RNS, respectively), including free radicals, and is augmented upon dysregulation of the production or removal mechanisms of the reactive species. Oxidative stress has been shown to be increased in the liver due to ethanol abuse, viral infection, and high-fat diets. In addition, the increase in the levels of ROS/RNS affects the initiation and progression of different hepatic pathologies [58][64][58,64]. As elaborated already, NF-κB activation is the first step in the canonical NLRP3 inflammasome activation, and ROS critically regulate the activity of the transcription factor NF-κB; for instance, the phosphorylation of Ser-276 of NF-κB p65 subunit is necessary for the positive transcription elongation factor b and is required for the expression of a subset of NF-κB-dependent genes [65]. However, there are other described mechanisms by which NF-κB can also be downregulated for ROS action. A specific cysteine residue in the p50 subunit of NF-κB is sensitive to oxidation by ROS. In particular, the oxidation of Cys-62 in the N-terminal DNA-binding domain of p50 inside the so-called Rel Homology domain [66] inhibits NF-κB DNA binding [67] and reduces NF-κB activation. Mechanistically, antioxidant treatment inhibits Ser-276 phosphorylation by regulating the ROS-dependent cAMP-dependent protein kinase catalytic subunit (PKAc) pathway, which contributes to NF-κB DNA-binding activity [68]. Accordingly, there exists reciprocal crosstalk between ROS production and NF-κB in the regulation of the inflammatory response, which affects the priming of the NLRP3 inflammasome.
Moreover, the role of ROS in the second step of canonical NLRP3 inflammasome activation has been extensively focstusdied. In this regard, thioredoxin (TRX)-interacting protein (TXNIP) has been identified as a binding protein of NLRP3 that facilitates its activation due to ROS production [69][69] (Figure 2). Most canonical NLRP3 activators (monosodium urate crystals, asbestos, alum, silica, imiquimod, or extracellular ATP) increase the production of ROS through nicotinamide adenine dinucleotide phosphate (NADPH). However, the NLRP3 inflammasome activation remains unaffected even after the genetic knockdown of some of the components of the NADPH oxidase complex (NOX1, NOX2, and NOX4), suggesting that different ROS sources could be activated during NLRP3 activation [70][71][72][70,71,72]. In that regard, mitochondria are the main cellular generators of ROS, and the NLRP3 inflammasome activation is dependent on ROS accumulation upon inhibition of autophagy and mitophagy (mechanisms by which the ROS production is controlled) [73]. AN increase in ROS results in the oxidation of TRX and its release from TXNIP, leaving TXNIP free for binding to the NLRP3, inducing its activation [69]. However, although ROS are important in the activation of NLRP3, it must also be accompanied by the efflux of intracellular K+ [69].