Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Hoe-Han Goh and Version 4 by Camila Xu.
Post-proline cleaving enzymes (PPCEs) are involved in various biological functions in diverse taxa of organisms including microbes, fungi, animals, and plants.
- prolyl endoprotease
- prolyl oligopeptidase
- protease
- protein engineering
1. Introduction
Peptidases, proteases, proteinases, or proteolytic enzymes break down peptide molecules, mostly by attacking peptide bonds via hydrolysis. Extensive protease studies found seven main catalytic types, which can be categorized by the nucleophiles for the proteolytic reaction with water molecules. The nucleophiles mainly comprise different amino acid residues, including aspartate, cysteine, glutamate, serine, and threonine, whilst another type contains metal ions (mostly zinc) at their active site, denoted as metalloproteases [1]. The seventh catalytic type performs intramolecular self-cleavage without water molecules using asparagine as a nucleophile [2]. Besides catalytic type, proteases can be further classified according to the peptide cleavage sites between the amino (N-) and carboxyl (C-) termini of proteins, such as aminopeptidase, carboxypeptidase, endopeptidase, or exopeptidase [3]. An oligopeptidase only cleaves peptides but not proteins. As of March 2022, there were 281 protein families in the MEROPS peptidase database, comprising 4431 peptidases with identifiers, which were classified into nine catalytic types aspartic (A, 324), cysteine (C, 1059), glutamic (G, 22), metallo (M, 1098), asparagine (N, 24), mixed (P, 54), serine (S, 1728), threonine (T, 105), and unknown/unclassifiable (U, 17).
Many PPCEs can be found in the MEROPS peptidase database from 17 families with different substrates and proline-cleavage specificities (Table 1).
Table 1. A non-exhaustive list of selected enzymes with post-proline cleaving activity based on records from the MEROPS peptidase database release 12.4.
| Family | Subfamily | Peptidase | UniProt/MEROPS ID | Specificity † | Substrate | Cleavage Site | Reference |
|---|---|---|---|---|---|---|---|
| G3 [1/1] |
G03.001 | Strawberry mottle virus glutamic peptidase | MER1365461 | 2/2 (100%) | Polyprotein | Peptide-Pro↓Ala/Lys-Peptide | [4] |
| M2 [4/7] |
M02.003 | Peptidyl-dipeptidase angiotensin-converting enzyme (ANCE) | Q10714/MER0001987 | 6/7 (86%) | Bradykinin |
Table 2. The post-proline cleaving enzymes that are reported in the different species discussed in this research.
| Taxa | Species (Common Name) | UniProt/PDB ID | MEROPS | Enzyme | Reference |
|---|---|---|---|---|---|
| Virus | Strawberry Mottle Virus | -/- | G03.001 | ||
| Vigna radiata | |||||
| DPP-II | |||||
| -4mβNA and -βNA dipeptides | |||||
| 7.5 | |||||
| 37 | |||||
| UV-Vis 520 nm | |||||
| [ | |||||
| 70 | |||||
| ] | |||||
| [ | |||||
| 74 | ] |
* -4mβNA: -4-methoxy-β-naphthylamide; -βNA: -beta-naphthylamide; ELISA: enzyme-linked immunosorbent assay; MS: mass spectrometry; MS/MS: tandem MS; RP-HPLC: reversed-phase high-performance liquid chromatography; NA: not available; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; ZGPpNA: benzyloxycarbonyl-Gly-Pro-p-nitroanalide.
Table 4.
The optimum conditions of selected recombinant PPCEs in substrate degradation.
| Source | Enzyme | Expression Host | Expression | Purification | Substrate | pH | T (°C) | Assay Detection | Reference | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fungi | Glutamic peptidase | [4] | |||||||||||||||||
| Peptide-Pro↓Phe/Tyr-Arg | [ | 5 | ] | ||||||||||||||||
| Archaea | Pyrococcus furiosus | Q51714/5T88 | S09.002 | POP | [46] | M02.006 | |||||||||||||
| Aspergillus niger | AN-PEP | Pischia pastoris GS115/pPIC9K | 28 °C/96 h | Ni-affinity chromatography | ZGPpNA | 4–5 | 35–40 | UV-Vis 410 nm | [91][95] | Bacteria | Aeromonus punctataAngiotensin-converting enzyme-2 | Q9BYF1/MER0011061 | 10/14 (71%) | Q9X6R4/3IUM | S09.001Angiotensin-2 | Peptide-Pro↓Phe | [6] | ||
| PEP | [ | 47 | |||||||||||||||||
| 35 | LC-MS/MS | [102] | M3 [8/11] |
M03.002 | Neurolysin | P42676/MER0001942 | 68/128 (53%) | Neurotensin | Peptide-Pro↓Tyr-Peptide | [7] | |||||||||
| M9D [1/2] |
M9D.002 | Proline-specific peptidyl-dipeptidase (Streptomyces) | - | 2/2 (100%) | Leu-Pro-Pro-Pro-Pro-Pro | Leu-Pro-Pro-Pro↓Pro-Pro | [8] | ||||||||||||
| ] | |||||||||||||||||||
| [ | 105 | ] | Flavobacterium meningosepticum | P27195/- | S09.UPA | POP | [48] | ||||||||||||
| Aspergillus oryzae | Prolyl aminopeptidase | Escherichia coli BL21/pET-28a(-) | 25 °C/12 h | Ni-affinity chromatography | L-Pro-pNA | 6.5–7.5 | 50 | - | [103][106] | Lactobacillus helveticus | O52071/- | M13.010 | PepO-3 | ||||||
| Bacteria | [ | 12] | M12 * [41/109] |
M12.164 | Lebetase | Q98995/MER0002591 | 6/14 (43%) | Bradykinin | Peptide-Pro↓Phe-Arg | [9] | |||||||||
| Meiothermus ruber H328 | A0A7C3HT26/- | S09.UPA | PEP/POP | [35] | |||||||||||||||
| Flavobacterium meningosepticum | Fm-POP | Wheat/pUC57 | - | - | Gluten peptide | 6 | 37 | HPLC, ELISA | [48] | M12.338 | BmooMPalpha-I (Bothrops sp.) | P85314/MER0104668 | 21/36 (58%) | Bradykinin | Peptide-Pro↓Phe-Arg | [10] | |||
| [ | 52 | ] | Mycobacterium tuberculosis | O07178/- | S09.036 | ||||||||||||||
| POP | [ | 31] | E. coli DH5α/pUC57 + Fmen | 37 °C/14 h | Geneclean III kit | ZGPpNA, gliadin, glutenin | - | Up to 90 | UV-Vis | M13 * [11/13] |
M13.005 | Oligopeptidase O3 (PepO) | MER0001059 | 3/7 (43%) | Beta-casein | Peptide-Pro↓Val/Ile-Peptide | [11 | ||
| ] | |||||||||||||||||||
| Myxococcus xanthus | Q9X5N2/2BKL | S09.076 | POP | [33] | M13.010 | Oligopeptidase O2 (PepO) | O52071/MER0004645 | 4/4 (100%) | Beta-casein | Peptide-Pro↓Val/Ile-Peptide | [12] | ||||||||
| Porphyromonas gingivalis | Q7MUW6/2D5L | S09.017 | PTP | [49] | M34 [3/3] |
M34.002 | Pro-Pro endopeptidase 1 (Clostridium difficile-type) | MER0494994 | 13/13 (100%) | Putative adhesin | Peptide-Asn-Pro↓Pro-Peptide | [13] | |||||||
| Serratia marcescens | O32449/1QTR | S33.001 | PAP | [50] | M34.003 | Pro-Pro endopeptidase 2 | MER0328914 | 3/3 (100%) | Putative s-layer protein | Peptide-Pro↓Pro-Peptide | |||||||||
| Sphingomonas capsulate | [ | Q9ZNM8/- | 14] | ||||||||||||||||
| S9.UPA | POP | [33] | M64 [1/1] |
M64.001 | IgA peptidase (Clostridium ramosum-type) | MER0016067 | 2/2 (100%) | Immunoglobulin IgA1 | Peptide-Pro↓Val-Peptide | ||||||||||
| Stenotrophomonas maltophilia | [ | A0A0U5BDB7/- | 15] | ||||||||||||||||
| S9.UPW | PEP | [51] | M72 [2/2] |
M72.002 | CpaA g.p. (Acinetobacter baumannii) | MER1365492 | 2/2 (100%) | Coagulation factor XII | Peptide-Pro↓Thr-Peptide | ||||||||||
| [ | 104 | ] | [ | 107 | ] | ||||||||||||||
| Meiothermus ruber H328 | MrPEP | E. coli BL21(DE3)/pET-28b | 37 °C/14 h | DEAE-cellulose and Phenyl-FF chromatography | ZGPpNA, suc-Ala-Pro-pNA, Gly-Pro-pNA, Pro-pNA | 9 | 60 | UV-Vis, FRETS-25Xaa libraries, LC-MS | [35][40] | ||||||||||
| Myxococcus xanthus | PEP | E. coli BL21(DE3)/pET-28b | 22 °C/16 h | Ni-affinity chromatography | Collagen peptide | 6–7 | 36–37 | UV-HPLC-MS/MS | [19][24] | ||||||||||
| Porphyromonas gingivalis | Prolyl tripeptidyl aminopeptidase | E. coli M15/pQE30-ptpA | 30 °C/18 h | Butyl-sepharose column | Fluorogenic substrate | 7.5 | 30 | UV-Vis | [49][53] | ||||||||||
| Sphaerobacter thermophiles | PEP | E. coli DE3/pET-15b | 37 °C/24 h | Ni-NTA resin spin column | ZGPpNA, gluten peptide | 6.6 | 63 | Enzymatic assay, MS | [105][108] | ||||||||||
| Sphingomonas capsulate | PEP | E. coli BL21(DE3)/pET-28b | 22 °C/16 h | DEAE and Blue sepharose chromatography | Chromogenic Gluten peptide | 6–7 | 36–37 | UV-HPLC-MS/MS | [19]Fungi | Aspergillus niger[ | A2QR21/7WAB16] | ||||||||
| [ | 24 | ] | S28.004 | AN-PEP | [38] | ||||||||||||||
| Insect | S6 [4/8] |
S06.001 | IgA1-specific serine peptidase (Neisseria-type) | MER0000278 | 6/6 (100%) | Immunoglobulin IgA1 | Peptide-Pro↓Xaa-Peptide | [17] | |||||||||||
| Aspergillus oryzae | A0A1S9DCM9/- | S28.004 | PEP | S06.007 | IgA1-specific serine peptidase type 1 (Haemophilus sp.) | MER0001759 | 3/3 (100%) | IgA1 chain C region | Peptide-Pro↓Thr-Peptide | [18] | |||||||||
| [ | 52 | ] | |||||||||||||||||
| Galerina marginata | H2E7Q8/5N4C | S09.077 | POP-B | [34] | S9 * [21/32] |
S09.001 | Prolyl oligopeptidase | P23687/MER0000392 | 50/59 (85%) | Alpha/beta-gliadin MM1 | Peptide-Pro↓Xaa-Peptide | [19] | |||||||
| Protozoa | Trypanosoma brucei | Q6HA27/- | S09.033 | POP | [53] | S09.002 | Prolyl oligopeptidase homologue (Pyrococcus-type) | MER0000398 | 2/3 (67%) | Z-Gly-Pro-NHPhNO2 | Z-Gly-Pro↓NHPhNO2 | [20] | |||||||
| Trypanosoma cruzi | Q71MD6/- | S09.033 | POP | [54] | S09.003 | Dipeptidyl-peptidase IV (eukaryote) | P27487/MER0000401 | 21/29 (72%) | C-X-C motif chemokine 10 | Val-Pro↓Leu-Peptide | [21 | ||||||||
| Insects | ] | ||||||||||||||||||
| Drosophila melanogaster | Q10714/2X91 | M02.003 | ANCE | [55] | S09.006 | Dipeptidyl aminopeptidase B (fungus) | P18962/MER0000405 | 5/7 (71%) | Alanyl/prolyl bond | Xaa-Ala/Pro↓Xaa | |||||||||
| Eurygaster integriceps | PEP | E. coli BL21(DE3)/pNYCO | 28 °C/16 h | Ni-affinity chromatography | ZGPpNA, wheat gluten | 7.5 | 37 | UV-Vis | [106][109] | ||||||||||
| Haematobia irritans exigua | ANCE | - | - | Lectin-affinity and ion-exchange chromatography |
Angiotensin 1, cholecystokinin-8 |
7.5 | 37[22] | ||||||||||||
| HPLC | Eurygaster integriceps (sunn pest) | E1U339/- | S09.001 | PEP | [56] | S09.007 | Fibroblast activation protein alpha subunit | Q12884/MER0000399 | 6/6 (100%) | Alpha-2-antiplasmin | Peptide-Pro↓Asn/Leu-Peptide | [ | |||||||
| Haematobia irritans (buffalo fly) | 23 | ] | |||||||||||||||||
| Q10715/ | - | M02.003 | PDP | [5] | S09.008 | Dipeptidyl peptidase 4 ( | |||||||||||||
| Animals | Aspergillus-type) | Haliotis discusMER0004504 | (abalone)6/8 (75%) | [Des-Arg]-bradykinin | Pro-Pro↓Gly-Peptide | [ | A0A1X9T5X9/6JYM | S09.UPA | PEP | [57]24] | |||||||||
| S09.009 | Dipeptidyl-peptidase 4 (bacteria-type 1) | Q5NMM8/MER0041840 | 3/5 (60%) | Gly-Pro-NHNap | Gly-Pro↓NHNap | [25 | |||||||||||||
| ] | |||||||||||||||||||
| Homo sapiens | P48147/3DDU | S09.001 | POP | [58][59] | S09.013 | Dipeptidyl-peptidase 4 (bacteria-type 2) | Q47900/MER0001423 | ||||||||||||
| [ | 5 | ] | [ | 10 | ] | P27487/1J2E | S09.00315/17 (88%) | Beta-casein | DPP-IV | [60]Tyr-Pro↓Phe-Peptide | [26] | ||||||||
| S09.017 | Prolyl tripeptidyl peptidase | Q7MUW6/MER0005196 | |||||||||||||||||
| 13/13 (100%) | P42785/3N2Z | S28.001 | Lysosomal PRCP | [Cystatin C | 61Ser-Ser-Pro↓Gly-Peptide | [27] | |||||||||||||
| ] | S09.018 | ||||||||||||||||||
| Dipeptidyl-peptidase 8 | Q6V1X1/MER0013484 | 8/10 (80%) | C-X-C motif chemokine 10 | Q9BYF1/1R42Val-Pro↓Leu-Peptide | M02.006 | ACE-2 | [62][28] | ||||||||||||
| S09.019 | Dipeptidyl-peptidase 9 | Q86TI2/MER0004923 | |||||||||||||||||
| Mus musculus | 13/15 (87%) | RU1 antigenic peptide | Val-Pro↓Tyr-Peptide | [29] | |||||||||||||||
| (mouse) | P42676/1I1I | M03.002 | Neurolysin (POP) | [63] | S09.033 | Prolyl oligopeptidase (zoomastigote) | Q4E132/MER0079308 | - | - | - | [30] | ||||||||
| Q9QUR6/- | S09.001 | POP | [64] | S09.036 | Rv0457C peptidase (Mycobacterium tuberculosis) | MER0003229 | 2/2 (100%) | Suc-Gly-Pro-NHMec | Peptide-Pro↓NHMec | [31] | |||||||||
| Bothrops sp. (snake) | P85314/3GBO | M12.338 | BmooMPalpha-I | [10] | S09.073 | Xaa-Pro dipeptidylpeptidase | D7UPN5/MER0195666 | 19/19 (100%) | Ala-Pro-NHPhNO2 | Xaa-Pro↓NHPhNO2 | [32] | ||||||||
| Sus scrofa (porcine) | P23687/1O6G | S09.001 | POP | [45] | S09.076 | Prolyl oligopeptidase (Myxococcus xanthus) | Q9X5N2/MER0005694 | 2/2 (100%) | Suc-Ala-Pro-NHPhNO2 | Peptide-Pro↓NHPhNO2 | [33] | ||||||||
| Plants | Arabidopsis thaliana | F4HSS5/- | S33.001 | PAP | [61] | S09.077 | Prolyl oligopeptidase B (Galerina marginata) | H2E7Q8/MER0325901 | 3/3 (100%) | Alpha-amanitin proprotein 1 | Peptide-Pro↓Ile/Trp-Peptide | [34] | |||||||
| Coffea arabica (coffee) | -/- | S09.001 | POP | [65] | S09.UPA | Subfamily S9A unassigned peptidases | - | 3/3 (100%) | Gly-Pro-NHPhNO2 | Gly-Pro↓NHPhNO2 | |||||||||
| Daucus carota (carrot) | -/- | S09.UPA | PEP | [66][35] | |||||||||||||||
| S15 [1/1] |
S15.001 | Xaa-Pro dipeptidyl-peptidase | Q02W78/MER0000443 | 8/10 (80%) | Gly-Pro-NHPhNO2 | Gly-Pro↓NHPhNO2 | [ | ||||||||||||
| Nepenthes sp. (pitcher plant) | A0A1V0DK55/- | 36 | ] | ||||||||||||||||
| U74.001 | Neprosin | [67] | S28 [2/3] |
S28.001 | Lysosomal Pro-Xaa carboxypeptidase | ||||||||||||||
| Secale cereale × Triticum turgidum subsp. durum (triticale) | P42785/MER0000446 | 11/11 (100%) | G9J616/- | S33.001 | PAPEndothelin B receptor-like protein 2 | Peptide-Pro↓Ala | [37] | ||||||||||||
| [ | 68 | ] | S28.004 | Acid prolyl endopeptidase (Aspergillus sp.) | A2QR21/MER0093133 | - | - | - | [38] | ||||||||||
| Spinacia oleracea (spinach) | P12301/- | U9G.029 | PEP | [69] | S33 [5/10] |
S33.001 | Prolyl aminopeptidase | P42786/MER0000431 | 4/6 (67%) | Consensus prolyl bond | Pro↓Xaa | [39] | |||||||
| Vigna radiata (mung bean) | -/- | S28.002 | DPP-II | [70] | S33.004 | Prolyl dipeptidase (Lactobacillus-type) | A8YWX2/MER0000437 | 17/18 (95%) | Pro-Gly | Pro↓Xaa | [40] | ||||||||
| S33.008 | Prolyl aminopeptidase 2 | P46547/MER0001367 | 9/10 (90%) | Pro-NHPhNO2 | Pro↓NHPhNO2 | [41] | |||||||||||||
| S37 [1/1] |
S37.001 | PS-10 peptidase | Q54408/MER0001350 | 3/3 (100%) | Transglutaminase precursor | Pepetide-Pro↓Asp-Peptide | [42] | ||||||||||||
| U9G [3/20] |
U9G.029 | Prolyl endopeptidase (Spinacia) | - | 3/3 (100%) | Oxygen-evolving enhancer protein 3, chloroplastic | Peptide-Pro↓Ile/Leu-Peptide | [43] | ||||||||||||
| U74 [1/1] |
U74.001 | Neprosin | - | 51/110 (46%) | Alpha/beta-gliadin MM1 | Peptide-Pro↓Xaa-Peptide | [44] |
↓: cleavage site; -: the linked amino acids; NA: not available; Xaa: any amino acid. † Specificity is based on the MEROPS specificity matrix showing the proportion of proline residue in the P1 cleavage site of all substrate cleavage records. Numbers in square brackets indicate the proportion of subfamily with records of post-proline cleavage (https://www.ebi.ac.uk/merops/cgi-bin/specsearch.pl) (accessed on 15 March 2022) with query XXX-XXX-XXX-PRO|XXX-XXX-XXX-XXX. * Overrepresentation (p < 0.001) is based on the hypergeometric test.
2. Molecular Structure and Biochemistry
2.1. Structural Studies
Protein structures of PPCEs have been studied across different taxa including bacteria, fungi, animals, and plants (Table 2). In this section, reswearchers selected some of the well-studied structures of PPCEs to depict their similarities and differences in hope of gaining insights on their regulation and molecular mechanisms for further investigations. The protein structures found in the PDB database are mostly solved through X-ray crystallography. The first crystal structure for a prolyl oligopeptidase (POP) was solved in 1998 at 1.4 Å resolution to reveal the α/β hydrolase fold and β-propeller domains with a catalytic triad of Ser-His-Asp [45]Note: ACE/ANCE: angiotensin-converting enzyme; DPP: dipeptidyl-peptidase; PAP: prolyl aminopeptidase; PDP: peptidyl-dipeptidase; PEP: prolyl endoproteinase; PepO: oligopeptidase O; POP: prolyl oligopeptidase; PRCP: Pro-Xaa carboxypeptidase; -: not available. Italic PDB ID indicates the crystal structure for the homology modeling in SWISS-MODEL.
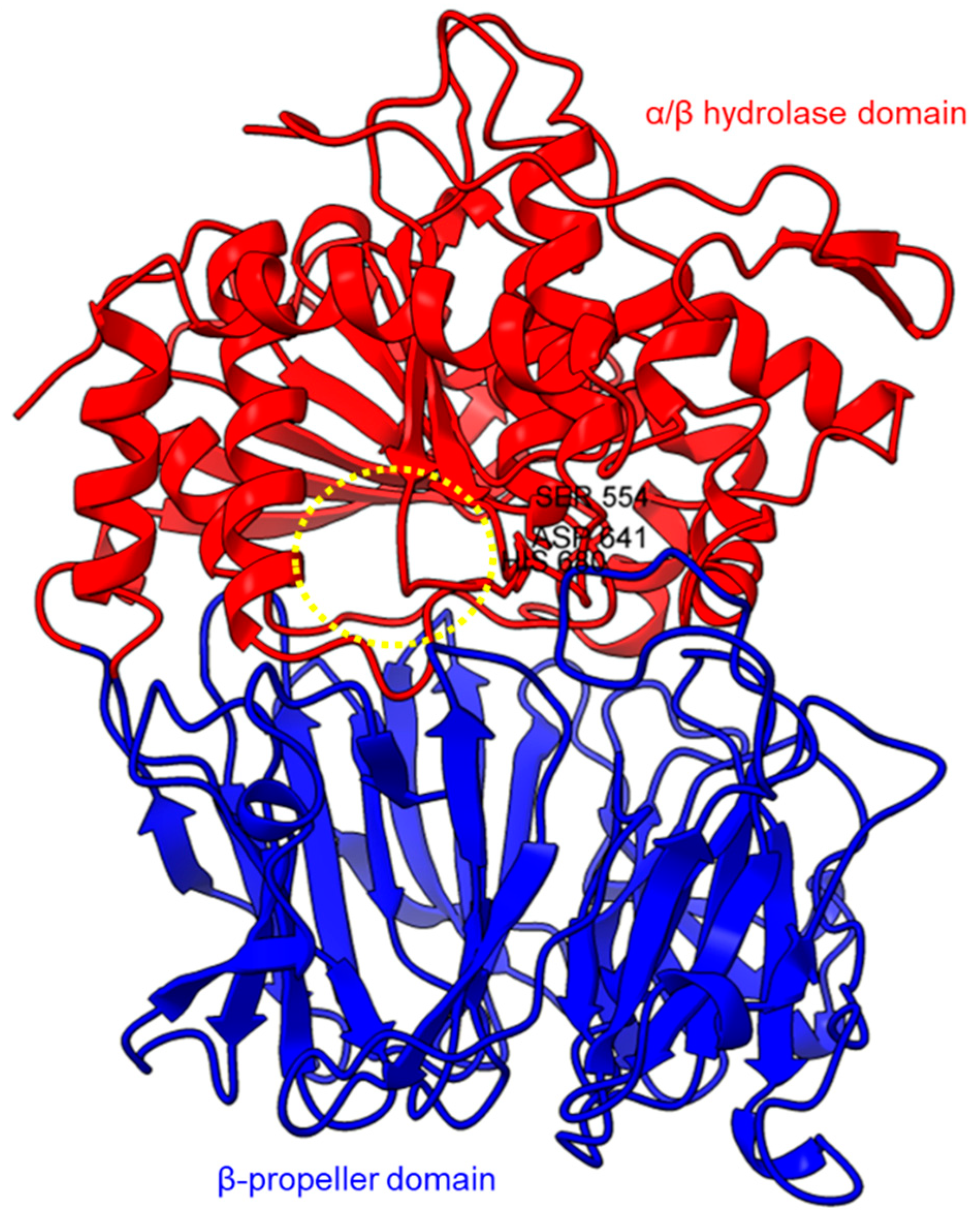
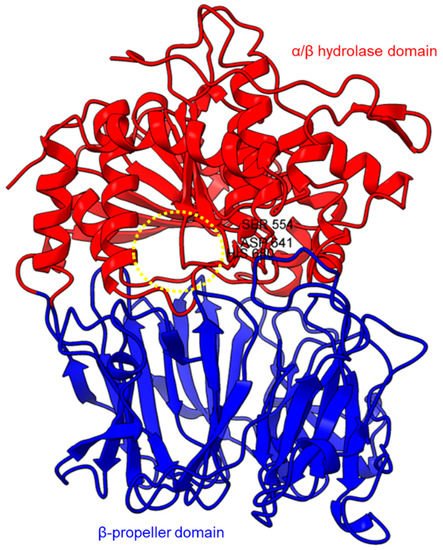
Based on the sequence alignments and structural models, POP proteins from the MEROPS S9 family have two conserved domains (Figure 1). The C-terminal catalytic α/β-hydrolase domain comprises an alpha/beta/alpha sandwich for protein cleaving, whereas the N-terminal β-propeller domain constitutes β-sheets that limit proteolysis to smaller substrates as a mechanism to avoid non-targeted digestion [33][47][71][38,51,75]. The electron microscopy of human PEP revealed the presence of a new side opening that was not observed in any of the crystallographic structures [71][75]. Two paths were identified using the CAVER algorithm, leading from the PEP active site to the outside solvent, one through the β-propeller and another one through a large side aperture into the catalytic domain [71][75].
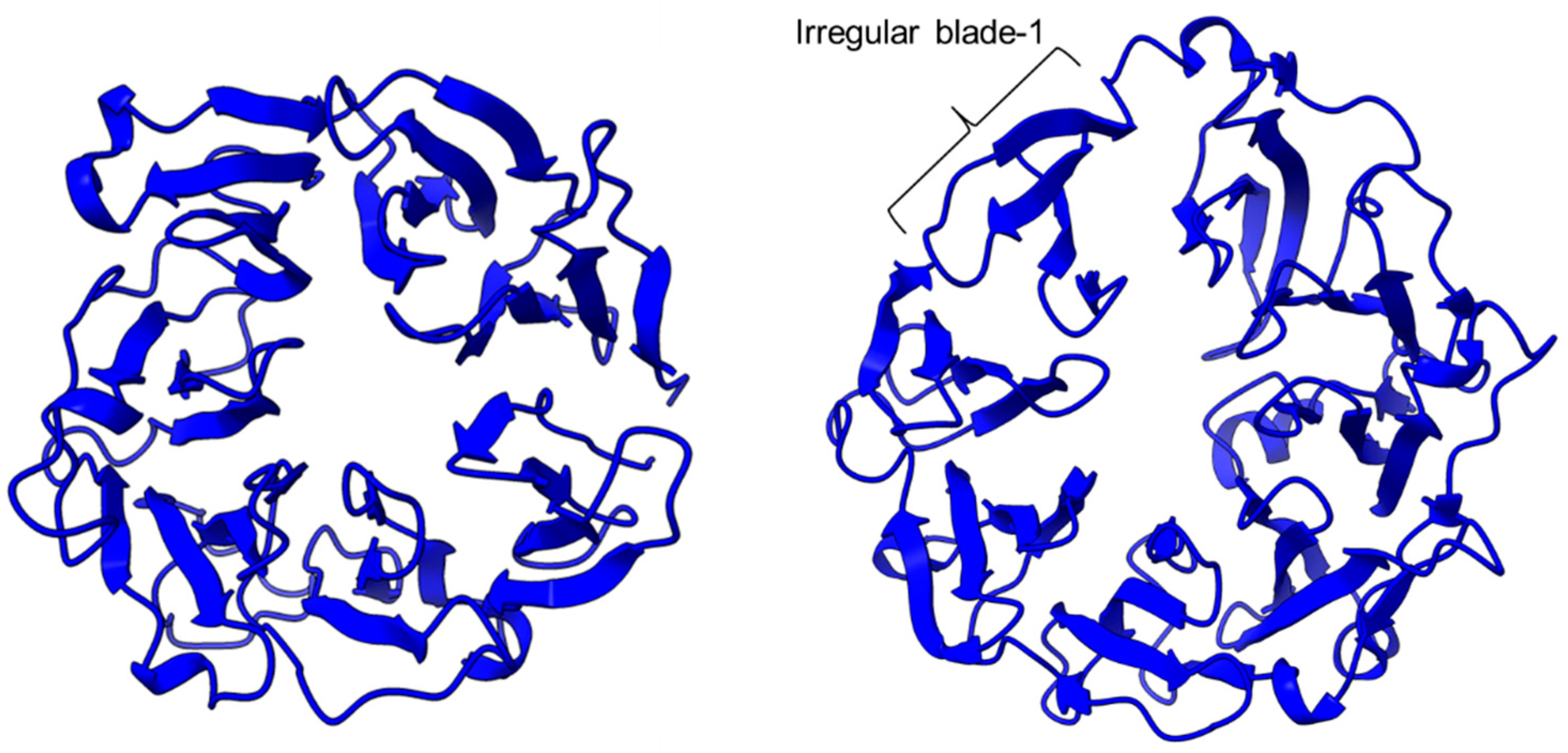
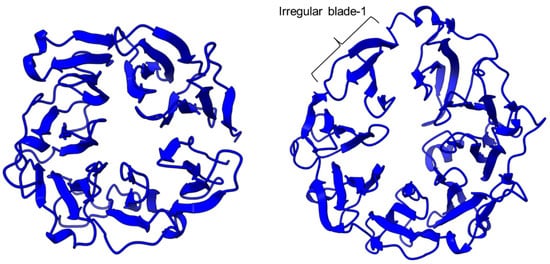
 PEP/POP from different species have different β-propeller sizes, thus conferring specificity to different sizes of substrates targeting proline residues that are usually resistant to protein cleavage by other peptidases [72][73][76,77]. For example, studies on Arabidopsis thaliana, Homo sapiens, and Sus scrofa (porcine) showed that each POP possesses a unique affinity toward different sizes of ligands despite high sequence and structural similarities [74][78]. Besides the pore size of β-propellers, the number of β-propeller blades also plays a role in the substrate specificity of PEP. For instance, the porcine POP (S9a, S09.001) has a seven-bladed β propeller structure that acts as a filtering gate to exclude large peptides from the active site [45][68], whereas the crystal structure of the human dipeptidyl-peptidase (DPP IV) (S9b, S09.003) shows a unique eight-bladed β-propeller (Figure 2). The irregular blade-1 on DPP-IV is hypothesized to allow substrate entry to the catalytic site without going through the gating filter compared to POP [60][63]. The difference in the β-propeller of POP and DPP-IV could have contributed to the substrate specificity of these two enzymes in which POP hydrolyses small peptides (<30 amino acids) at the C-terminal of proline residues, while DPP-IV cleaves dipeptides at the penultimate proline residue. As compared to the β-propeller domains, the α/β-hydrolase catalytic domain is mostly conserved.
PEP/POP from different species have different β-propeller sizes, thus conferring specificity to different sizes of substrates targeting proline residues that are usually resistant to protein cleavage by other peptidases [72][73][76,77]. For example, studies on Arabidopsis thaliana, Homo sapiens, and Sus scrofa (porcine) showed that each POP possesses a unique affinity toward different sizes of ligands despite high sequence and structural similarities [74][78]. Besides the pore size of β-propellers, the number of β-propeller blades also plays a role in the substrate specificity of PEP. For instance, the porcine POP (S9a, S09.001) has a seven-bladed β propeller structure that acts as a filtering gate to exclude large peptides from the active site [45][68], whereas the crystal structure of the human dipeptidyl-peptidase (DPP IV) (S9b, S09.003) shows a unique eight-bladed β-propeller (Figure 2). The irregular blade-1 on DPP-IV is hypothesized to allow substrate entry to the catalytic site without going through the gating filter compared to POP [60][63]. The difference in the β-propeller of POP and DPP-IV could have contributed to the substrate specificity of these two enzymes in which POP hydrolyses small peptides (<30 amino acids) at the C-terminal of proline residues, while DPP-IV cleaves dipeptides at the penultimate proline residue. As compared to the β-propeller domains, the α/β-hydrolase catalytic domain is mostly conserved.
 There is a recent report of the Aspergillus niger PEP (AN-PEP) structure solved through an x-ray crystallography [38][43]. AN-PEP belongs to the peptidase family S28 of Pro-Xaa carboxypeptidase, which is in the same SC clan with family S9 sharing the same catalytic site residues (Ser-Asp-His) and likely the same evolutionary origin. The AN-PEP structure consists of 17 α-helices, 10 310helices, and 10 β-strands. In contrast to the POP of peptidase family S9, the β-propeller domain is replaced by a helical SKS domain (Figure 3) that is stabilized by three disulfide bonds. The catalytic pocket of AN-PEP is located between the catalytic α/β hydrolase domain and the SKS domain. AN-PEP is capable of digesting large substrates, unlike POP with a substrate limit of less than 30 amino acids. The substrate specificity could be due to the differences in the catalytic pocket structure formed between the α/β hydrolase and SKS domains in AN-PEP, compared to the β-propeller domain of S9 PEP/POP.
There is a recent report of the Aspergillus niger PEP (AN-PEP) structure solved through an x-ray crystallography [38][43]. AN-PEP belongs to the peptidase family S28 of Pro-Xaa carboxypeptidase, which is in the same SC clan with family S9 sharing the same catalytic site residues (Ser-Asp-His) and likely the same evolutionary origin. The AN-PEP structure consists of 17 α-helices, 10 310helices, and 10 β-strands. In contrast to the POP of peptidase family S9, the β-propeller domain is replaced by a helical SKS domain (Figure 3) that is stabilized by three disulfide bonds. The catalytic pocket of AN-PEP is located between the catalytic α/β hydrolase domain and the SKS domain. AN-PEP is capable of digesting large substrates, unlike POP with a substrate limit of less than 30 amino acids. The substrate specificity could be due to the differences in the catalytic pocket structure formed between the α/β hydrolase and SKS domains in AN-PEP, compared to the β-propeller domain of S9 PEP/POP.
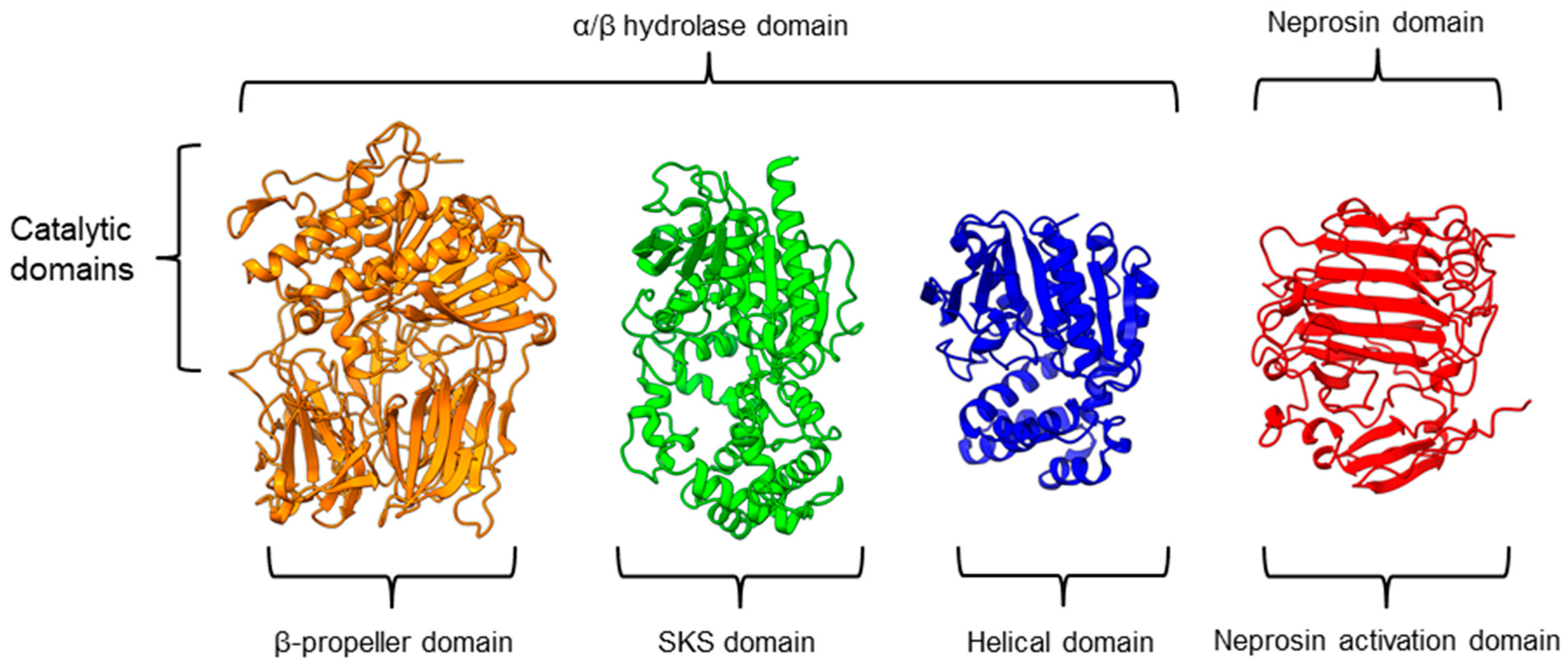
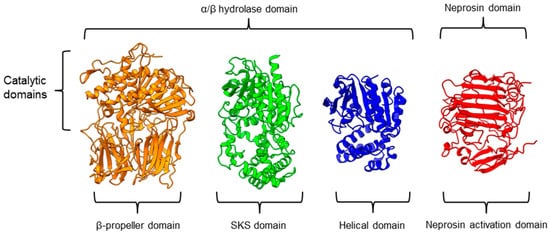 Another family of PPCEs is the prolyl aminopeptidase (PAP) from the peptidase family S33, which can be found in apricot seeds [19][24] and cabbage leaves [75][79]. Prolyl aminopeptidase (S33.001) is an exopeptidase that catalyzes the removal of the proline residue at the N-terminal of a peptide [50][54]. Like peptidase families S9 and S28, the catalytic α/β hydrolase domain is present (Figure 3). However, it has a catalytic triad of Ser113, His296, and Asp 268 with a consensus sequence of GXSXG around the catalytic serine residue. The second unannotated helices domain comprises six α-helices that act as a cap to block the N-terminal of the pre-cleavage (P1) proline, thus explaining the exopeptidase catalytic mechanism. PAPs are categorized based on their functioning structures: 30–35 kDa monomers (S33.001) are found exclusively in bacteria, while 100–370 kDa multimers (S33.008) have been reported in bacteria, fungi, and plants. Interestingly, a biochemically characterized plant PAP from triticale is more like the monomeric PAPs than the multimeric form [73][77].
On the other hand, a protease from the carnivorous tropical pitcher plants known as neprosin with PPC activity was classified as an unknown peptidase family (U74). There is a recent neprosin structure–function analysis based on the AlphaFold2 modeling that suggests neprosin belongs to the glutamic peptidase family with two glutamic acid residues as the catalytic dyad [76][80]. Unlike the S9, S28, or S33 prolyl proteases, neprosin does not have an α/β hydrolase domain, SKS domain, or β-propeller domain. Instead, neprosin has two uncharacterized domains, namely the neprosin activation domain and the neprosin domain (Figure 3). The neprosin domain is proposed to be the catalytic domain due to the absence of the neprosin activation domain in the active enzyme either in the native or recombinant neprosins [44][77][49,81]. Intriguingly, the neprosin domain comprises predominantly β-sheets, which form two antiparallel six- and seven-stranded β-sheets with an overall β-sandwich structure.
In comparison, all the PPCEs from family S9, S28, S33, and neprosin have a two-domains structure. The α/β hydrolase domain and neprosin domain act as the catalytic domain, whereas the second domain (SKS, β-propeller, helical, and neprosin activation domains) appears to be involved in other functions such as substrate size limitation in S28 and S33 PEP, potential protein–protein interactions for S9 POP [78][82], and enzyme maturation/activation in the case of neprosin. Interestingly, two-domain structures are observed in these different families of PPCEs from diverse taxa, despite a lack of homology in amino acid sequences. This suggests a convergent functional and structural evolution.
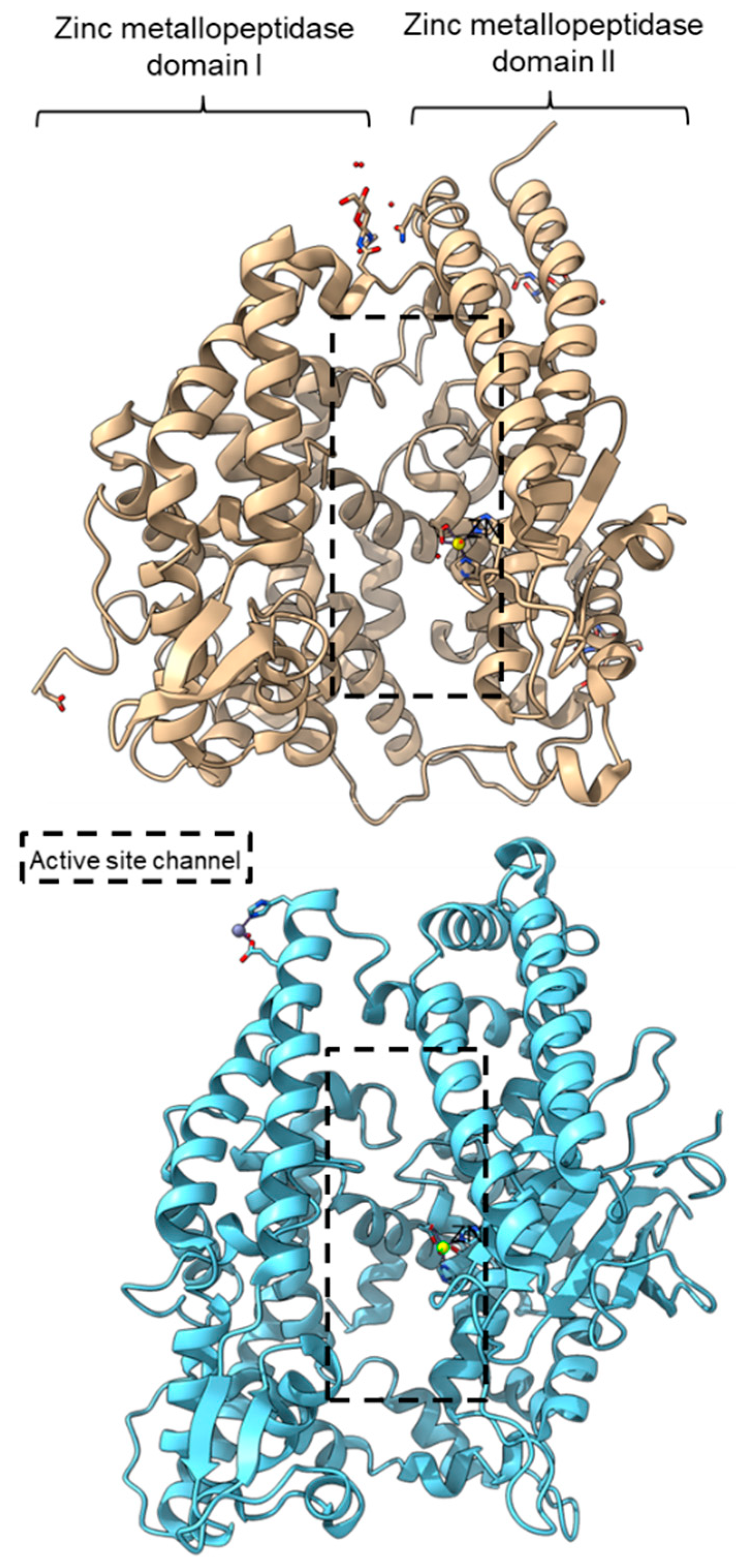
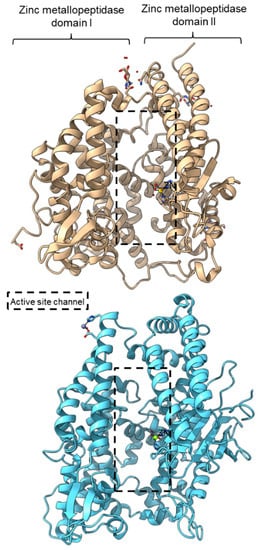
PPC activity has also been reported in a few members of zinc metallopeptidase families (M2, M3, M9D, M12, M13, M34, M64, and M72). Peptidyl-dipeptidase ANCE (M02.003), angiotensin-converting enzyme-2, ACE2 (M02.006), and neurolysin (M03.002) comprise two domains annotated as zinc metallopeptidase domain I and domain II [62][63][79][65,66,83]. An active channel with a bound zinc ion is located in between the two active helices domains connected by small secondary structures (Figure 4). One of the structural differences between ACE2 and neurolysin is the number of the strand of the β-sheet on domain II, where ACE2 has a three-stranded β-sheet, while neurolysin has a five-stranded β-sheet (Figure 4). Furthermore, the more flexible loop structures on neurolysin could have contributed to its wide substrate specificity [63][66].
Another family of PPCEs is the prolyl aminopeptidase (PAP) from the peptidase family S33, which can be found in apricot seeds [19][24] and cabbage leaves [75][79]. Prolyl aminopeptidase (S33.001) is an exopeptidase that catalyzes the removal of the proline residue at the N-terminal of a peptide [50][54]. Like peptidase families S9 and S28, the catalytic α/β hydrolase domain is present (Figure 3). However, it has a catalytic triad of Ser113, His296, and Asp 268 with a consensus sequence of GXSXG around the catalytic serine residue. The second unannotated helices domain comprises six α-helices that act as a cap to block the N-terminal of the pre-cleavage (P1) proline, thus explaining the exopeptidase catalytic mechanism. PAPs are categorized based on their functioning structures: 30–35 kDa monomers (S33.001) are found exclusively in bacteria, while 100–370 kDa multimers (S33.008) have been reported in bacteria, fungi, and plants. Interestingly, a biochemically characterized plant PAP from triticale is more like the monomeric PAPs than the multimeric form [73][77].
On the other hand, a protease from the carnivorous tropical pitcher plants known as neprosin with PPC activity was classified as an unknown peptidase family (U74). There is a recent neprosin structure–function analysis based on the AlphaFold2 modeling that suggests neprosin belongs to the glutamic peptidase family with two glutamic acid residues as the catalytic dyad [76][80]. Unlike the S9, S28, or S33 prolyl proteases, neprosin does not have an α/β hydrolase domain, SKS domain, or β-propeller domain. Instead, neprosin has two uncharacterized domains, namely the neprosin activation domain and the neprosin domain (Figure 3). The neprosin domain is proposed to be the catalytic domain due to the absence of the neprosin activation domain in the active enzyme either in the native or recombinant neprosins [44][77][49,81]. Intriguingly, the neprosin domain comprises predominantly β-sheets, which form two antiparallel six- and seven-stranded β-sheets with an overall β-sandwich structure.
In comparison, all the PPCEs from family S9, S28, S33, and neprosin have a two-domains structure. The α/β hydrolase domain and neprosin domain act as the catalytic domain, whereas the second domain (SKS, β-propeller, helical, and neprosin activation domains) appears to be involved in other functions such as substrate size limitation in S28 and S33 PEP, potential protein–protein interactions for S9 POP [78][82], and enzyme maturation/activation in the case of neprosin. Interestingly, two-domain structures are observed in these different families of PPCEs from diverse taxa, despite a lack of homology in amino acid sequences. This suggests a convergent functional and structural evolution.
PPC activity has also been reported in a few members of zinc metallopeptidase families (M2, M3, M9D, M12, M13, M34, M64, and M72). Peptidyl-dipeptidase ANCE (M02.003), angiotensin-converting enzyme-2, ACE2 (M02.006), and neurolysin (M03.002) comprise two domains annotated as zinc metallopeptidase domain I and domain II [62][63][79][65,66,83]. An active channel with a bound zinc ion is located in between the two active helices domains connected by small secondary structures (Figure 4). One of the structural differences between ACE2 and neurolysin is the number of the strand of the β-sheet on domain II, where ACE2 has a three-stranded β-sheet, while neurolysin has a five-stranded β-sheet (Figure 4). Furthermore, the more flexible loop structures on neurolysin could have contributed to its wide substrate specificity [63][66].
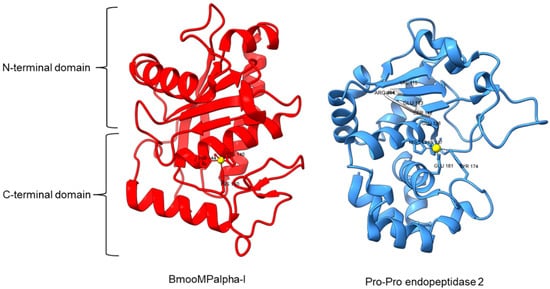
 Metallopeptidase M12 BmooMPalpha-I (M12.338, 3gbo) with a molecular mass of 22.6 kDa has conserved features of P-I class snake venom metallopeptidases, namely the five-stranded β-sheet, four long helices, and a short α-helix near N-terminal stabilized by three disulfide bridges [10][15]. The zinc ion is bound to three histidine residues: His140, His144, and His150 (Figure 5). Additionally, Pro-Pro endopeptidase 1, PPEP-1 (M34.002, 5a0p), and Pro-Pro endopeptidase 2, PPEP-2 (M34.003, 6fpc) shared a very similar structure with an α/β N-terminal domain (NTD) comprising twisted four-stranded β-sheet and three α-helices (α1-α3), and an α C-terminal domain (CTD) with four α-helices (α5-α8) [14][80][19,84]. The active site of Pro-Pro endopeptidase 2 is located at the α4-helix that separates NTD and CTD domains. A zinc ion is bound to the catalytic base Glu138, two histidine residues (His137 and His141) from α4-helix, Tyr174 from α5/α6-helices, and Glu181 from α6-helix (Figure 5). The four amino acid substrate loops (S-loops) in PPEP-1 (GGST) and PPEP-2 (SERV) are shown to contribute to the difference in their substrate specificities [14][19]. Therefore, peptidases with proline-cleaving activity are not confined to the serine peptidase family alone. The advent of accurate ab initio protein structure prediction from sequences will reveal more peptidases conferring PPC activity via different catalytic mechanisms with different structures and provide a further understanding of the structure-function of the PPCEs.
Metallopeptidase M12 BmooMPalpha-I (M12.338, 3gbo) with a molecular mass of 22.6 kDa has conserved features of P-I class snake venom metallopeptidases, namely the five-stranded β-sheet, four long helices, and a short α-helix near N-terminal stabilized by three disulfide bridges [10][15]. The zinc ion is bound to three histidine residues: His140, His144, and His150 (Figure 5). Additionally, Pro-Pro endopeptidase 1, PPEP-1 (M34.002, 5a0p), and Pro-Pro endopeptidase 2, PPEP-2 (M34.003, 6fpc) shared a very similar structure with an α/β N-terminal domain (NTD) comprising twisted four-stranded β-sheet and three α-helices (α1-α3), and an α C-terminal domain (CTD) with four α-helices (α5-α8) [14][80][19,84]. The active site of Pro-Pro endopeptidase 2 is located at the α4-helix that separates NTD and CTD domains. A zinc ion is bound to the catalytic base Glu138, two histidine residues (His137 and His141) from α4-helix, Tyr174 from α5/α6-helices, and Glu181 from α6-helix (Figure 5). The four amino acid substrate loops (S-loops) in PPEP-1 (GGST) and PPEP-2 (SERV) are shown to contribute to the difference in their substrate specificities [14][19]. Therefore, peptidases with proline-cleaving activity are not confined to the serine peptidase family alone. The advent of accurate ab initio protein structure prediction from sequences will reveal more peptidases conferring PPC activity via different catalytic mechanisms with different structures and provide a further understanding of the structure-function of the PPCEs.

 For peptidase family S28, it is suggested that the difference in the catalytic pocket located between α/β hydrolase and the SKS domain could confer substrate specificity based on the recent AN-PEP crystal structure [38][43]. The catalytic pocket formed in AN-PEP is wide-open compared to the lysosomal Pro-Xaa carboxypeptidase (PRCP) (S28.001) and dipeptidyl peptidase II, DPP7 (S28.002) with a volume-limited catalytic pocket that will affect the recognition of substrates with different sizes [61][64]. However, Glu88, Pro205, Trp374, and Trp460 residues are conserved across PEP, PRCP, and DPP7 despite differences in the volumes of catalytic pocket.
Different from S9 and S28 families, the S33 family has exopeptidase properties whereby it cleaves the proline residues at the N-terminal of a peptide. Prolyl aminopeptidase (PAP) from Serratia marcescens (S33.001) has a similar α/β hydrolase domain and another smaller helical domain made up of six helices, which are smaller than the SKS domain on S28. The smaller domain of prolyl aminopeptidase is believed to contribute to the exopeptidase specificity of S33 prolyl protease as the specificity hydrophobic pocket is formed on the helical domain [50][54]. Two mutagenesis studies on prolyl aminopeptidase from Serratia marcescens concur that residues Phe139, Tyr149, and Glu204 play an important role in the substrate recognition [82][83][86,87]. Phe139, Tyr149, and Phe236 in the hydrophobic pocket together with Glu204 and Glu232 are involved in electrostatic interactions. The proline recognition is completed by four electrostatic interactions and the insertion of substrate into the hydrophobic pocket of prolyl aminopeptidase [82][86]. Substrate acetylation could help in the orientation of the substrate in the active cleft for efficient hydrolysis [84][88]. The catalytic mechanism of peptidase family S28 and S33 could be like that of S9 prolyl endopeptidase, as they shared a similar classical serine catalytic triad of Ser-Asp-His.
Other than the serine peptidases, a glutamic peptidase was reported with PPC activity and a catalytic dyad comprising two catalytic glutamic acid residues [4][9]. In silico analysis of neprosin found a high structural similarity to strawberry mottle virus glutamic peptidase (SMoV peptidase, G03.001) with an overlapping catalytic dyad [76][80]. This shed light on the possible catalytic mechanism of neprosin. The mature neprosin has a putative accessible active cleft formed by the β-sandwich structure for substrate binding. The catalytic mechanism of glutamic peptidase G3 could be like that of the Gln and Glu catalytic dyad in the G1 family because the main catalytic Glu residue of glutamic peptidase G1 is found to be conserved in the G3 family [85][89].
For peptidase family S28, it is suggested that the difference in the catalytic pocket located between α/β hydrolase and the SKS domain could confer substrate specificity based on the recent AN-PEP crystal structure [38][43]. The catalytic pocket formed in AN-PEP is wide-open compared to the lysosomal Pro-Xaa carboxypeptidase (PRCP) (S28.001) and dipeptidyl peptidase II, DPP7 (S28.002) with a volume-limited catalytic pocket that will affect the recognition of substrates with different sizes [61][64]. However, Glu88, Pro205, Trp374, and Trp460 residues are conserved across PEP, PRCP, and DPP7 despite differences in the volumes of catalytic pocket.
Different from S9 and S28 families, the S33 family has exopeptidase properties whereby it cleaves the proline residues at the N-terminal of a peptide. Prolyl aminopeptidase (PAP) from Serratia marcescens (S33.001) has a similar α/β hydrolase domain and another smaller helical domain made up of six helices, which are smaller than the SKS domain on S28. The smaller domain of prolyl aminopeptidase is believed to contribute to the exopeptidase specificity of S33 prolyl protease as the specificity hydrophobic pocket is formed on the helical domain [50][54]. Two mutagenesis studies on prolyl aminopeptidase from Serratia marcescens concur that residues Phe139, Tyr149, and Glu204 play an important role in the substrate recognition [82][83][86,87]. Phe139, Tyr149, and Phe236 in the hydrophobic pocket together with Glu204 and Glu232 are involved in electrostatic interactions. The proline recognition is completed by four electrostatic interactions and the insertion of substrate into the hydrophobic pocket of prolyl aminopeptidase [82][86]. Substrate acetylation could help in the orientation of the substrate in the active cleft for efficient hydrolysis [84][88]. The catalytic mechanism of peptidase family S28 and S33 could be like that of S9 prolyl endopeptidase, as they shared a similar classical serine catalytic triad of Ser-Asp-His.
Other than the serine peptidases, a glutamic peptidase was reported with PPC activity and a catalytic dyad comprising two catalytic glutamic acid residues [4][9]. In silico analysis of neprosin found a high structural similarity to strawberry mottle virus glutamic peptidase (SMoV peptidase, G03.001) with an overlapping catalytic dyad [76][80]. This shed light on the possible catalytic mechanism of neprosin. The mature neprosin has a putative accessible active cleft formed by the β-sandwich structure for substrate binding. The catalytic mechanism of glutamic peptidase G3 could be like that of the Gln and Glu catalytic dyad in the G1 family because the main catalytic Glu residue of glutamic peptidase G1 is found to be conserved in the G3 family [85][89].
Figure 1. Crystal structure of a porcine POP (PDB ID: 1QFM). In red is the α/β hydrolase catalytic domain, whereas the β-propeller domain is highlighted in blue. Catalytic triad residues: Ser554, Asp641, and His680 are labeled. The yellow circle indicates a probable accessible path discovered in an electron microscopy study [71][75].
Figure 2. The β-propeller structure of a porcine prolyl oligopeptidase POP (PDB id: 1QFM, left) and a human dipeptidyl peptidase DPP-IV (PDB ID: 1J2E, right). The labeled irregular blade-1 of the eight-blade β-propeller structure could contribute to different substrate specificity between POP and DPP-IV.
Figure 3. Representative protein structures from the MEROPS peptidase family S9 (porcine muscle prolyl oligopeptidase, PDB ID: 1QFM), S28 (Aspergillus niger prolyl endoprotease, PDB ID: 7WAB), S33 (Serratia marcescens prolyl aminopeptidase, PDB ID: 1QTR), and U74 (AlphaFold2 model of N. × ventrata neprosin, NvNpr), from left to right. The three-dimensional protein structures are oriented with their catalytic domains on top.
Figure 4. Crystal structures of the MEROPS metallopeptidase M02.006 angiotensin-converting enzyme-2, ACE2 (top, PDB ID: 1R42), and M03.002 neurolysin (bottom, PDB ID: 1I1I).
Figure 5. Crystal structures of MEROPS metallopeptidase M12.338 BmooMPalpha-I (left, PDB ID: 3GBO) and M34.003 Pro-Pro endopeptidase 2 (right, PDB ID: 6FPC) with substrate loop (S-loop) SERV in light gray. The zinc ions are shown as yellow spheres.
2.2. Molecular Mechanisms of PPCEs
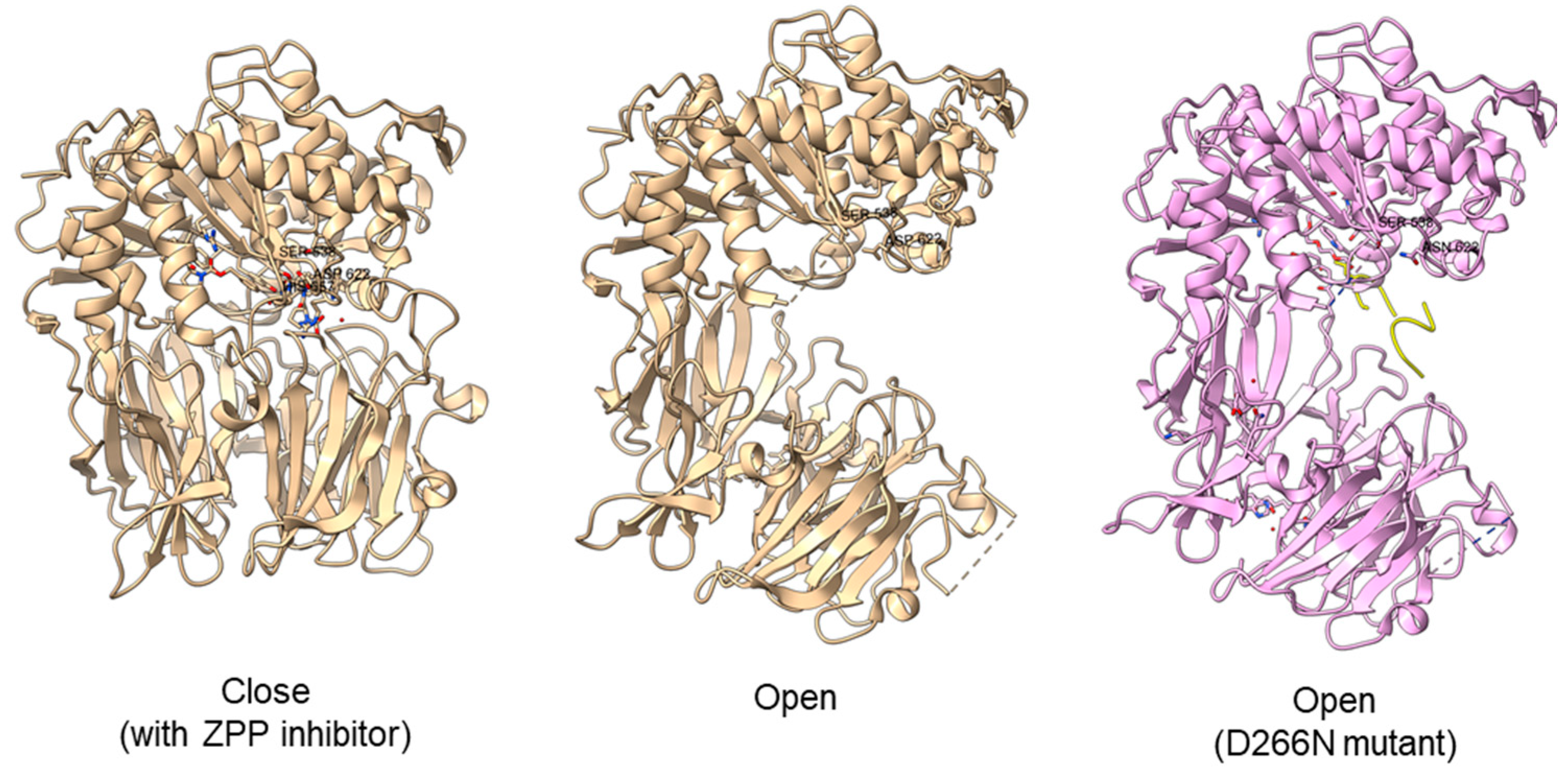
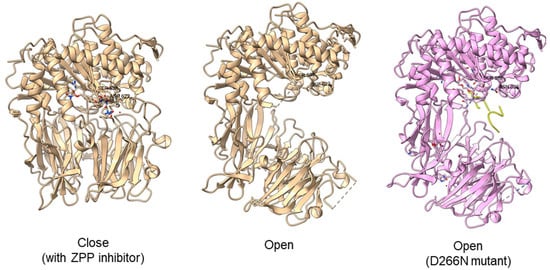
Typically, PEP/POP from the clan SC of family S9 comprises a catalytic triad with the following order, Ser-Asp-His with the catalytic Ser lies in between the Gly-X-Ser-X-Gly motif [45][68]. However, there are also “nonclassical” serine proteases such as the Ser/His/Glu, Ser/His/His, and Ser/Glu/Asp triads, Ser/Lys and Ser/His dyads, and even peptidases with a single serine catalyst [81][85]. For S9 PEP, two major hypotheses on substrate binding mechanism had been proposed. The initial hypothesis is that the substrate enters the filtering gate and the central channel formed by the β-propeller structure before reaching the active site. This theory is supported by Sphingomonas capsulate PEP, as it failed to digest the 33-mer substrate (LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF) effectively with a diameter larger than the 4 Å channel formed by the β-propeller domain. However, this theory cannot explain why S9 PEP from Myxococcus xanthus and Flavobacterium meningosepticum can digest the 33-mer substrate in the same study. Later, it is proposed that substrate binding with the β-propeller domain induces conformation changes, causing the domain to move and expose the active site. This theory is supported by the open state of Sphingomonas capsulate PEP structure, where an opening of 30 Å is observed between the α/β hydrolase and β-propeller domain [33][38]. Another study obtained an open state of Aeromonus punctata PEP and proposed an induced-fit mechanism as the conformational changes in the PEP can be induced by adding substrate and inhibitor [47][51]. The open state is achieved when the substrate H2H3 is added, while a closed state occurs after the binding of the Z-prolyl-prolinal (ZPP) inhibitor with PEP (Figure 6). HP35 substrate with over 30 amino acids can enter the active site but is not cleaved, as the catalytic residue is not in an active conformation in the open state, whereas the catalytic pocket with the active catalytic triad conformation in a close state is too small for HP35. Recently, a molecular simulation of Pyrococcus furiosus POP (S09.002) shows that the conformational change is modulated by the “latching loop” mechanism and is essential for the catalysis mechanism of POP, especially for the loop containing catalytic histidine residue (H592) [46][50]. The conformation of the histidine loop during the shifting of open to close state allows H592 to move closer to the active site for the catalytic triad formation. Chloride ion binding is required for the loop movement with subsequent activation of the POP peptidase catalysis.Figure 6. Crystal structures of Aeromonas punctata PEP (ApPEP). From left to right: close-state wild type ApPEP with ZPP inhibitor (PDB ID: 3IVM), open-state wild type ApPEP (PDB ID: 3IUL), mutant ApPEP with D266N mutation in an open state (PDB ID: 3IUR) induced by two H2H3 substrates (yellow). The catalytic triad (Ser538, Asp622, and His657) are labeled. However, the loops containing histidine catalytic residue (His657) are missing in the open-state crystal structures as they are unresolved in electron density maps and are inherently flexible.
2.3. Biochemical Studies of PPCEs
The native (Table 3) and recombinant proteins (Table 4) of PPCEs have been reported for different species, mainly microbes and animals. For the assays of PPC activity, Z-Gly-Pro-pNA (ZGPpNA) and other chromogenic substrates are commonly used [86][90]. PPCEs specifically cleave at the carboxyl-end of proline residue of the chromogenic substrates, releasing chromogenic pNA (p-nitroanilide) or βNA (β-naphthylamide), which can be detected spectrophotometrically at a wavelength of 410 nm and 520 nm, respectively. Due to their specificity, these chromogenic substrates are often used in the functional characterization and activity measurement of PPCEs. Furthermore, proline-rich gliadin is also a common substrate to investigate the capability of certain PPCEs in gluten detoxification. The fragments of gliadin hydrolysates can be detected through ELISA, Western blot, or immunoblot with specific antibodies. To ascertain the protein cleavage sites, a liquid chromatography–mass spectrometry (LC-MS) approach can be taken to identify the digested peptide sequences. Specific amino acid preferences of targeted substrate sequences are curated in the MEROPS database with a collection of cleavage sites (Table 1). The substrate specificity will influence the molecular interactions between the protease and substrate at the active site of the protein with a role in biological functions [87][91]. Experiments to identify the substrate specificity of PPCEs utilize LC-MS [77][81] and fluorogenic substrates [88][92]. One study used the spectrofluorometer to determine the fluorometric Z-Gly-Pro- pNA peptide at the excitation (340 nm) and emission wavelengths (410 nm) [88][92]. In this researchtudy, the POP enzyme was shown to form strong interactions with no more than six residues from positions P4, P3, and P2 (N-side) and P1′ and P2′ (C-side) regardless of the substrate length [88][92]. PPCEs can hydrolyze proteins with different preferences of amino acids adjacent to the proline residues. For example, the recombinant PEP from a Gram-negative thermophile, Meiothermus ruber H328, shows a stringent preference of residues next to proline but a greater flexibility in preferences of the second and third residues near proline [35][40]. In a study using 3375 synthetic peptides as substrates of internally quenched fluorogenic probes (IQFPs), about 74% showed high favorability towards proline at the Xaa position [89][93]. Both cationic and anionic charged residues such as Arg/Lys/Asp/Glu are unfavorable at the subsequent position after proline. The anionic residues are more tolerated in position P2 compared to P3 with strong preferences toward Leu/Ile/Arg/His and low preferences towards Asp/Glu at P3 positions [90][94]. Therefore, PPCEs have a certain degree of specificity towards their substrates based on their amino acid sequences. Studies on both the native and recombinant PPCEs, mainly PEP/POP, showed similar optimum temperatures with pH 4–6, except for Eurygaster integriceps, Haliotis discus, Sus sucrofa, Porphyromonas gingivalis, and Nepenthes × ventrata (Table 3 and Table 4). Most of the native protein studies were based on Aspergillus niger with pH 4–5 and 37 °C. Nearly all recombinant protein characterization studies used Escherichia coli as a cloning and expression host apart from Pichia pastoris and wheat (Table 4). Some PPCEs exhibited different preferences for different substrate lengths and specificity with a broad range of optimum temperatures and pH levels. For instance, a POP (S09.001) cleaves after the C-terminal proline residue in peptide substrates with less than 30 amino acids. In addition to the PPC activity that cleaves at two conserved proline residues of α-amanitin pro-peptide, POP-B from Galerina marginata (GmPOPB, S09.077) has a unique transpeptidation activity that catalyzes macrocyclization of a 25-mer peptide to form a monocyclic octapeptide [34][39]. A PEP from Aspergillus oryzae (S28.004) exhibits a similar PPC activity at an optimal pH of 2.5 with the capability of digesting much larger substrates such as intact casein [91][95]. Like S28 PEP, neprosin with a lower molecular mass (~30 kDa) shows PPC activity at pH 2.5 but with no limitation of substrate size [77][81]. Prolyl aminopeptidase (S33.001) from family S33 has a proline-specific exopeptidase activity that cleaves proline at the N-terminal of a peptide [50][54]. All PPCEs showed the ability to cleave proline-rich substrates. These studies show the potential of PPCEs from different species for industrial applications with various conditions of pH and temperatures.Table 3.
The optimum conditions of selected native PPCEs in substrate degradation.
| Source | Enzyme | Substrate * | pH | T (°C) | Method to Detect Digested Substrate * | Reference | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fungus | ||||||||||||||||
| Aspergillus niger | PEP | Beer hordein | - | - | ELISA RIDASCREEN® Gliadin competitive | [92][96] | ||||||||||
| 4–5 | - | Gluten ELISA assay | [93][97] | |||||||||||||
| - | 14 | Antibody-based competitive enzyme-linked immunosorbent assay | [94][98] | |||||||||||||
| 4 | 37 | Gluten ELISA assay | [95][99] | |||||||||||||
| 4.6 | 37 | Western blot, monoclonal antibody-based competition assays | [96][100] | |||||||||||||
| 4–5 | 37 | SDS-PAGE, Western blot, ELISA, HPLC, MS, | [97][101] | |||||||||||||
| Wheat flour, gliadin | 4 | 40 | ELISA, immunoblot (anti-PEP-I, anti-PEP-II, and anti-HMW-GS antibodies), RP-HPLC | [98][7] | ||||||||||||
| N-glycosidase | 4–4.5 | 37 | MALDI-TOF MS | [99][102] | ||||||||||||
| ZGPpNA | 4 | 37 | Turbidity (protein-polyphenol haze) | [100][103] | ||||||||||||
| 4–5 | 40–50 | UV-Vis 410 nm | [101][104] | |||||||||||||
| Insect | ||||||||||||||||
| Eurygaster integriceps (sunn pest) | PEP | ZGPpNA | 8 and 10 | 24–34 | UV-Vis 410 nm | [56][6] | ||||||||||
| Animal | ||||||||||||||||
| Mus musculus | POP | Suc-Gly-Pro-AMC | 4.2 | 30 | Fluorescence plate reader | [64][67] | ||||||||||
| Plants | ||||||||||||||||
| Animals | Nepenthes × ventrata | Neprosin | α-gliadin | 2.5 | 37 | SDS-PAGE, turbidity monitored at 595 nm, MS/MS | [44 | |||||||||
| Haliotis discus (abalone) | PEP | E. coli BL21(DE3) | 18 °C/15 h | ] | [ | 49] | ||||||||||
| Ni-affinity chromatography | Collagen peptide | 6 | 20 | HPLC-MS | [ | 33 | ] | [ | 38] | Spinacia oleracea PSII membranes |
PEP | Co-purified 23 kDa and 18 kDa proteins | 6 | 37 | Densitogram | [69] |
| Sus sucrofa | POP | [ | E. coli Top10/pBAD | 37 °C/4–6 h | 73 | ] | ||||||||||
| DEAE and Blue sepharose chromatography | ZGPpNA -AMC | 5–8 | 30 | UV-Vis | [ | 107 | ] | [ | 110] | |||||||
| Plant | ||||||||||||||||
| Nepenthes × ventrata | Neprosin | E. coli Arctic Express/pET-28a(+) | 16 °C/16 h | Ni-affinity chromatography | HeLa cell protein | 2.5 | 37–50 | MS | [77][81] | |||||||
| Histone | 2.5 | 37 | LC-MS/MS | [108][111] |
Note: AMC: 7-amido-4-methylcoumarin; DEAE: diethylaminoethyl; ELISA: enzyme-linked immunosorbent assay; Hb: hemoglobin; HPLC: high-performance liquid chromatography; L-Pro-pNA: L-arginyl-L-proline 4-nitroanilide; MS: mass spectrometry; Ni-NTA: Ni2+-nitrilotriacetate; UV-Vis: ultraviolet-visible spectroscopy; ZGPpNA: Z-Gly-Pro-4-nitroanilide; -: not available.