Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Conner Chen and Version 2 by Conner Chen.
Mitochondria are the power plants of the cell responsible not only for the generation of cellular energy required for myriad functions but also are important hubs for metabolism, reactive oxygen species (ROS) generation, and Ca2+ homeostasis. Inflammasomes are a group of intracellular multicomplexes located in the cytosol which detect PAMPs and DAMPs and produce the activation, maturation, and release of pro-inflammatory cytokines (including IL-1β and IL-18).
- NLRP3 inflammasome
- mitochondria
- steatohepatitis
1. Introduction
Alcoholic (ASH) and nonalcoholic steatohepatitis (NASH) are two of the most common causes of chronic liver disease worldwide. ASH is caused by chronic alcohol consumption, while NASH is associated with dietary habits and obesity and linked to insulin resistance and type 2 diabetes [1][2]. Although ASH and NASH differ in their etiology, both pathologies are characterized by the excess of fat deposition and lipotoxicity leading to inflammation, hepatocellular ballooning, and fibrogenesis [3][4]. Despite significant advances in the identification of players mediating the transition from steatosis to steatohepatitis, unfortunately the therapeutic options for ASH and NASH are limited, due to our incomplete understanding of the molecular mechanisms contributing to ASH and NASH development.
Mitochondria are the power plants of the cell responsible not only for the generation of cellular energy required for myriad functions but also are important hubs for metabolism, reactive oxygen species (ROS) generation, and Ca2+ homeostasis. Associated with the role of energy production, mitochondria consume large amounts of molecular oxygen in the respiratory chain and, hence, are critical players in the onset of oxidative stress, which is accentuated by the disturbance of mitochondrial function related to hepatocyte injury in ASH and NASH [5]. As oxidative stress reflects the imbalance between oxidants and antioxidants defense, the impairment in the levels and/or activity of mitochondrial antioxidant strategies, including disruption of the GSH redox cycle, enhances the accumulation of ROS and increases the susceptibility of the cells to inflammatory cytokines [6].
Furthermore, mitochondrial dysfunction can activate the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome, leading to inflammation [7]. Inflammasomes are multiprotein immune complexes that are activated in response to pathogen-associated and danger-associated molecular patterns (PAMPs and DAMPs), such as lipopolysaccharide (LPS) and cholesterol crystals [8]. The activation of NLRP3 inflammasome followed by caspase-1 activation allows the release of proinflammatory cytokines, such as IL-1β and IL-18, into the extracellular space that promote inflammatory cell death (pyroptosis), leading to an increase in liver inflammation, steatosis, and fibrosis [9].
2. Overview of Mitochondria and NLRP3 Inflammasome Interplay
Inflammasomes are a group of intracellular multicomplexes located in the cytosol which detect PAMPs and DAMPs and produce the activation, maturation, and release of pro-inflammatory cytokines (including IL-1β and IL-18) [10][11]. Among the family of inflammasomes, the best characterized are NLRP1, NLRP3, NLRC4, and absent in melanoma 2 (AIM2) [12]. NLRP3, the best-studied member of the inflammasome family, is a multicomplex formed by NLRP3, adaptor apoptosis speck protein (ASC), and pro-caspase-1 [13]. Activation of the NLRP3 inflammasome can occur through two signal steps [8][14]. The priming signal is provided by microbial components (LPS) or endogenous cytokines (tumor necrosis factor alpha, TNF-α; IL-1β), followed by the binding to toll-like receptors (such as TLR4), leading to the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which in turn contributes to the transcription and translation of NLRP3. Then, NLRP3 undergoes post-translational modifications, resulting in its activation. The activation of the NLRP3 inflammasome leads to the subsequent activation of caspase 1, which can then cleave inactive pro-IL-1β and pro-IL-18 into active IL-1β and IL-18, respectively. The activation of caspase-1 also cleaves gasdermin D (GSDMD) into a N-terminal fragment, which transfers to the cell membrane for specific interactions with lipids. Cells undergo morphological changes, including plasma membrane rupture and pore formation, loss of ionic gradients, and osmosis resulting in cellular swelling due to water entry, and are finally lysed, causing the release of intracellular pro-inflammatory molecules (IL-1β and IL-18), which subsequently stimulate secondary cytokine production [15]. The signals for the inflammasome activation include extracellular stimuli such as adenosine triphosphate (ATP), pore-forming toxins, RNA viruses, cholesterol crystals, uric acid, and amyloid β [8][14]. There are three hypotheses regarding the activation of NLRP3 inflammasome: (a) the destabilization of the ion outflow (K+, Ca2+, and H+), creating holes in the cell membrane; (b) the release of ROS by damaged mitochondria; and (c) the rupture of lysosomes that produce the leak of the lysosomal enzyme cathepsin B [8]. Mitochondria are the main source of ROS (mtROS) (Figure 1).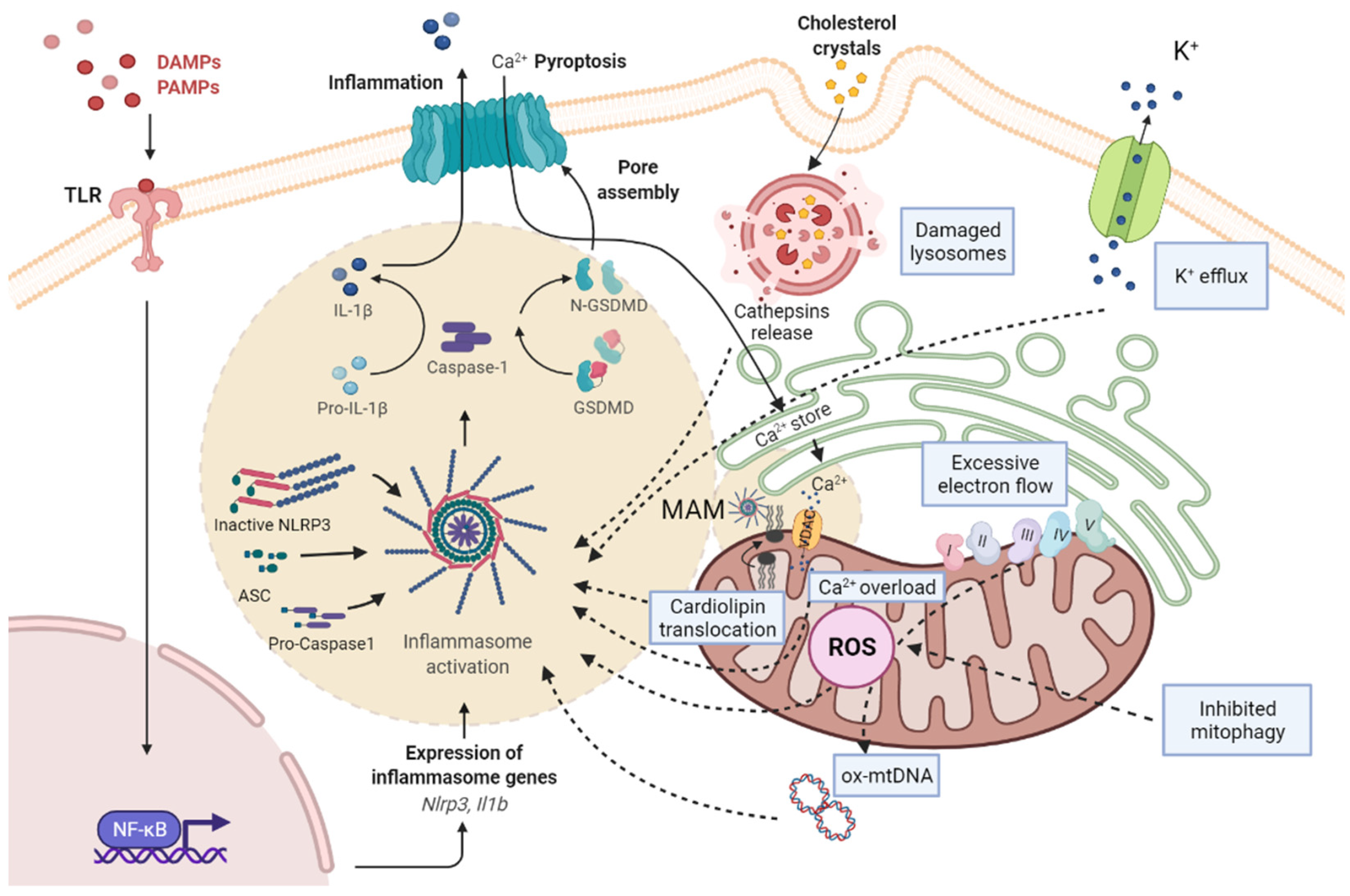
Figure 1. Scheme of the pathways associated with NRLP3 inflammasome activation and mitochondrial dysfunction. PAMPs and DAMPs are recognized by TLR, as priming signal, and activate NF-κB signaling pathway, promoting the transcription of pro-IL-1β and NLRP3, while an activation signal initiates the formation of NLRP3/ASC/pro-caspase-1 complex. Subsequently, the active form of caspase-1 cleaves pro-IL-1β to mature IL-1β and GSDMD into a N-terminal fragment (N-GSDMD). N-GSDMD produces pores in the membrane, allowing the release of IL-1β into the extracellular space. The mechanisms involved in NLRP3 activation include: the destabilization of the ion outflow (K+ efflux/Ca2+ influx) creating holes in the cell membrane; the release of ROS by damaged mitochondria produced by excessive electron flow; an inhibited mitophagy and mtDNA oxidation; and the release of cathepsins by damaged lysosomes. The NLRP3 inflammasome complex activation is located in MAMs, where cardiolipin translocation and Ca2+ influx by VDAC occurs, and facilitates the assembly of the NLRP3 inflammasome. ASC, adaptor apoptosis speck protein; DAMPS, damage-associated molecular patterns; GSDMD, gasdermin D; IL-1β, interleukin-1β; MAM, mitochondria-associated membranes; NF-kB, nuclear factor kappa B; NLRP3, NLR family pyrin domain containing 3; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species; TLR, toll-like receptor.
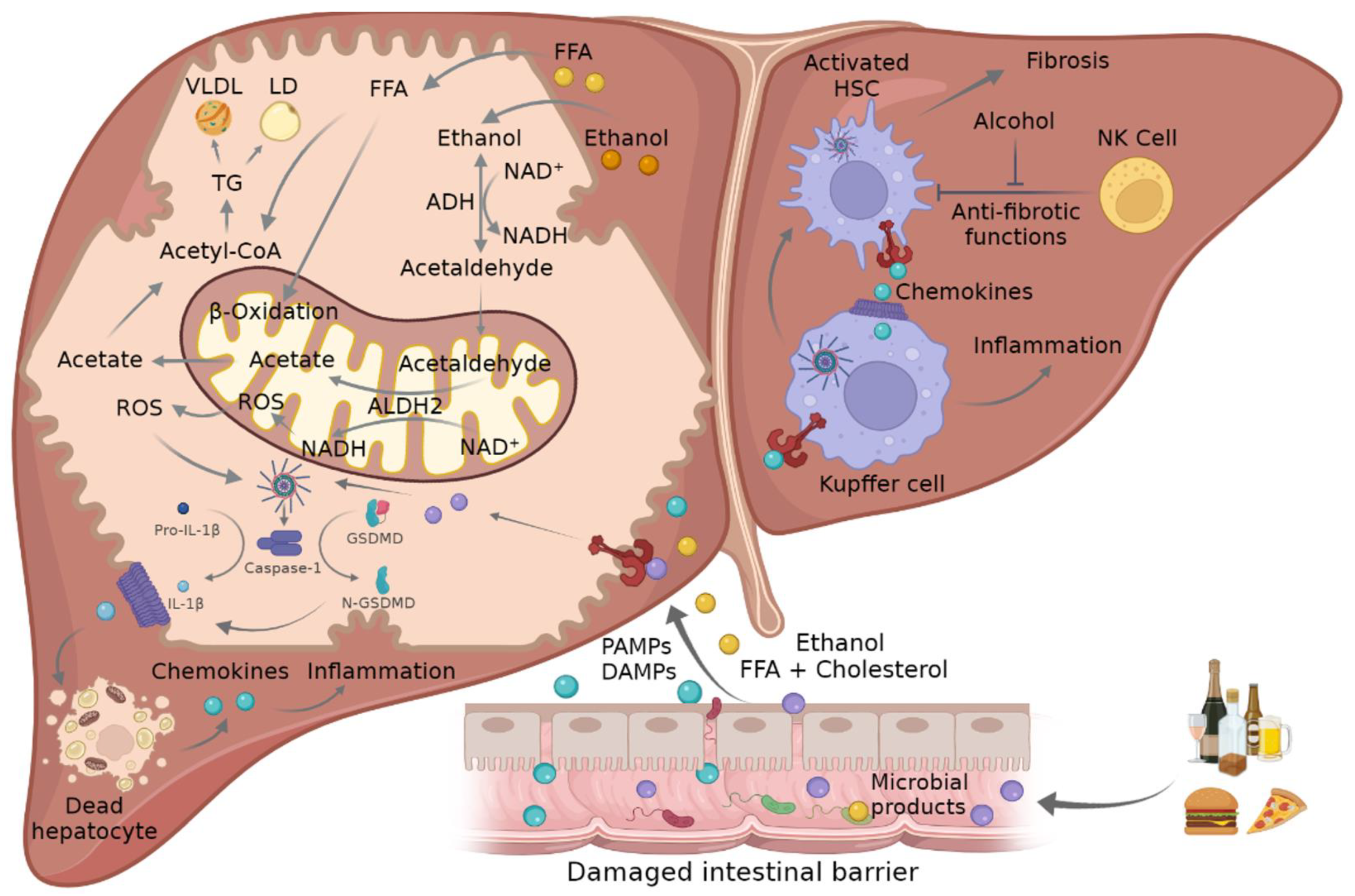
Figure 2. Mechanism of crosstalk between mitochondria and inflammasome in ASH and NASH. Ethanol consumption and FFA- and cholesterol-enriched diets can lead to damaged intestinal barrier where PAMPs and DAMPs such as FFA or microbial products go through the disrupted tight junctions and promote NLRP3 inflammasome activation in liver cells. This triggers the activation of caspase-1, which mediates the cleavage of pro-IL-1β and GSDMD into their mature forms, which in turn promote hepatocellular death and the attraction and activation of KCs and HSCs, leading to inflammation and fibrosis. The metabolism of ethanol and FFA in hepatocytes induces an increment in ROS in the mitochondria that in turn triggers inflammasome activation. ADH, alcohol dehydrogenase; ALDH2, acetaldehyde dehydrogenase 2; ASC, adaptor apoptosis speck protein; DAMPS, damage-associated molecular patterns; FFA, free fatty acid; GSDMD, gasdermin D; HSC, hepatic stellate cells; IL-1β, interleukin-1β; KC, Kupffer cells; NADH/NAD+, oxidized and reduced nicotinamide adenine dinucleotide ratio; NLRP3, NLR family pyrin domain containing 3; NK, natural killer cells; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle.
References
- Argemi, J.; Ventura-Cots, M.; Rachakonda, V.; Bataller, R. Alcoholic-Related Liver Disease: Pathogenesis, Management and Future Therapeutic Developments. Rev. Esp. Enferm. Dig. 2020, 112, 869–878.
- Cariello, M.; Piccinin, E.; Moschetta, A. Transcriptional Regulation of Metabolic Pathways via Lipid-Sensing Nuclear Receptors PPARs, FXR, and LXR in NASH. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1519–1539.
- Pelusi, S.; Cespiati, A.; Rametta, R.; Pennisi, G.; Mannisto, V.; Rosso, C.; Baselli, G.; Dongiovanni, P.; Fracanzani, A.L.; Badiali, S.; et al. Prevalence and Risk Factors of Significant Fibrosis in Patients With Nonalcoholic Fatty Liver Without Steatohepatitis. Clin. Gastroenterol. Hepatol. 2019, 17, 2310–2319.
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis Progression in Nonalcoholic Fatty Liver vs. Nonalcoholic Steatohepatitis: A Systematic Review and Meta-Analysis of Paired-Biopsy Studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.
- Robertson, D.J.; Yang, V.W.; Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647.
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharmacol. 2014, 5, 151.
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–226.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Torres, S.; Brol, M.J.; Magdaleno, F.; Schierwagen, R.; Uschner, F.E.; Klein, S.; Ortiz, C.; Tyc, O.; Bachtler, N.; Stunden, J.; et al. The Specific NLRP3 Antagonist IFM-514 Decreases Fibrosis and Inflammation in Experimental Murine Non-Alcoholic Steatohepatitis. Front. Mol. Biosci. 2021, 8, 715765.
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832.
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687.
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36.
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the Nlrp3 Inflammasome. Front. Immunol. 2019, 10, 2538.
- Zhang, Y.; Dong, Z.; Song, W. NLRP3 Inflammasome as a Novel Therapeutic Target for Alzheimer’s Disease. Signal. Transduct. Target. Ther. 2020, 5, 37.
- Rodríguez-Antonio, I.; López-Sánchez, G.N.; Uribe, M.; Chávez-Tapia, N.C.; Nuño-Lámbarri, N. Role of the Inflammasome, Gasdermin D, and Pyroptosis in Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. Hepatol. 2021, 36, 2720–2727.
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nuñez, S.; Nuño-Lámbarri, N.; Enrich, C.; García-Ruiz, C.; Fernández-Checa, J.C. Cholesterol Enrichment in Liver Mitochondria Impairs Oxidative Phosphorylation and Disrupts the Assembly of Respiratory Supercomplexes. Redox Biol. 2019, 24, 101214.
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833.
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124.
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553.
- Hong, T.; Chen, Y.; Li, X.; Lu, Y. The Role and Mechanism of Oxidative Stress and Nuclear Receptors in the Development of NAFLD. Oxidative Med. Cell Longev. 2021, 2021, 6889533.
- Quan, Y.; Xin, Y.; Tian, G.; Zhou, J.; Liu, X. Mitochondrial ROS-Modulated MtDNA: A Potential Target for Cardiac Aging. Oxidative Med. Cell Longev. 2020, 2020.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414.
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323.
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than Just a Housekeeper. Trends Cell Biol. 2009, 19, 81–88.
- Yu, J.W.; Lee, M.S. Mitochondria and the NLRP3 Inflammasome: Physiological and Pathological Relevance. Arch. Pharmacal. Res. 2016, 39, 1503–1518.
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-ΚB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910.
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174.
- Hoque, R.; Vodovotz, Y.; Mehal, W. Therapeutic Strategies in Inflammasome Mediated Diseases of the Liver. J. Hepatol. 2013, 58, 1047–1052.
More