Ubiquitination is reversed by the activity of deubiquitinase enzymes (DUBs). About 100 human DUBs are known, and they are divided into seven major families: the cysteine proteases of the USP (ubiquitin-specific proteases), UCH (ubiquitin C-terminal hydrolases), OTU (ovarian tumor), MJD (Machado-Joseph domain-containing proteases), MINDY (motif interacting with the Ub-containing novel DUB family), and ZUFSP (zinc finger with the UFM1-specific peptidase domain protein) families and the Zn-dependent metalloproteases of the JAMM (JAB1/MPN/MOV34 domain-associated) family. DUBs play a role in seemingly every biological process and are central to many human pathologies, thus rendering them very desirable and challenging therapeutic targets. Despite significant drug discovery efforts, only approximately 15 chemical probe-quality small molecule inhibitors have been reported, hitting just 6 of about 100 known DUBs.
- deubiquitinase (DUB)
- ubiquitin (Ub)
- inhibitor
1. Introduction
1.1. Deubiquitinase Enzymes: Classification and Activity Regulation
Ubiquitination is reversed by the activity of deubiquitinase enzymes (DUBs) (Figure 1). About 100 human DUBs are known, and they are divided into seven major families: the cysteine proteases of the USP (ubiquitin-specific proteases), UCH (ubiquitin C-terminal hydrolases), OTU (ovarian tumor), MJD (Machado-Joseph domain-containing proteases), MINDY (motif interacting with the Ub-containing novel DUB family), and ZUFSP (zinc finger with the UFM1-specific peptidase domain protein) families and the Zn-dependent metalloproteases of the JAMM (JAB1/MPN/MOV34 domain-associated) family [4][13][14][15][16][4,16,17,18,19]. The USP family is the largest, with 58 constituents. The members of each family are related through structurally homologous catalytic domains but can vary greatly by the presence of insertions, deletions, and additional domains serving diverse functions. Cysteine protease DUBs utilize a catalytic triad or dyad to facilitate a nucleophilic attack and hydrolysis of the scissile bond connecting the C-terminal Gly-76 of Ub to a substrate or other Ub of a poly-Ub chain. Contrarily, zinc-dependent metalloprotease DUBs (metalloDUBs) utilize a coordinated zinc ion and an activated, nucleophilic water molecule for catalysis [4]. DUBs are essential for maintaining Ub homeostasis by processing Ub precursors (UBC, UBB, UBA52, and UBA80) [17][20] and unconjugated chains [14][17]; recycling Ub at the proteasome [18][21]; and deubiquitinating various targets. Additionally, they function as interpreters and editors of a highly complex code. E3-DUB complexes are common and couple conjugation and deconjugation to facilitate chain editing and fine-tuning of the molecular output [19][22]. In the rare case of A20 (an OTU family member), both E3 and DUB activities are encoded into a single multifunctional enzyme [20][23]. The factors dictating DUB activity and substrate specificity include the structure of the catalytic domain, the presence of additional domains for substrate recruitment or Ub binding [21][22][23][24,25,26], alternative splicing [24][27], allostery [25][28], oligomerization [26][29], PTMs [27][30], and subcellular localization [4]. Importantly, not all DUBs are catalytically active. Approximately 10% are pseudo-enzymes currently found in the USP, OTU, JAMM, and MINDY families [28][31]. Pseudo-DUBs are critical components of large macromolecular complexes or as allosteric modulators of active DUBs. Most famously, PSMD7, a pseudo-DUB, dimerizes with the active DUB PSMD14. This complex is a critical component of the 19S proteasome regulatory particle [29][30][31][32,33,34]. Without detectable activity, pseudo-DUBs are most readily identified by sequence and structural similarity to catalytically competent homologs. DUBs can discriminate the linkage type, chain length, modifier (Ub or Ubl), and/or the substrate to which Ub is conjugated [32][35]. Furthermore, they can remove poly-Ub chains in different modes: iteratively at the distal end (exo-cleavage), from within a chain (endo-cleavage), or all at once (en bloc-cleavage) [33][36]. Members of the USP family are generally linkage-nonspecific and often referred to as promiscuous [21][34][35][24,37,38]; however, using tool reagents, including small molecule compounds and activity-based probes, aour group and others have revealed surprising linkage-dependent processing activities for USP9X [36][39] and USP7 [37][40], two USPs previously believed to be nonspecific. Whether or not other members of the USP family have a similar specificity remains to be explored. Contrarily, members of the OTU, MINDY, ZUFSP, and JAMM families tend to be linkage-specific [4][14][22][35][38][39][40][41][42][4,17,25,38,41,42,43,44,45]. For OTUD1, a C-terminal Ub-interacting motif (UIM) endows specificity for K63 linkages by forming a proximal Ub-binding site (S1’) to orient the scissile bond across the catalytic site [22][25]. The structural determinants of the linkage specificity can also be leveraged for chain length specificity. The five Ub-binding sites of the catalytic domain of MINDY1/2 render strict K48 linkage specificity and the ability to sense the poly-Ub chain length to modulate the exo- (<6 Ub) and endo-cleavage (>5 Ub) modes [43][46]. UCH DUBs preferentially cleave Ub from small or unstructured C-terminal leaving groups. This represents a sort-of substrate specificity that results from an active site crossover loop limiting the size of ubiquitinated substrates able to occupy the catalytic cleft [44][47].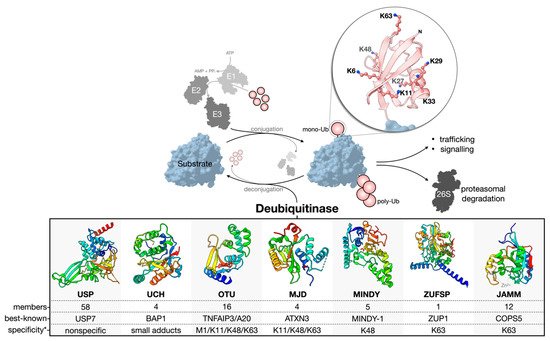
1.2. Challenges and Missteps When Studying DUBs at the Cellular Level
Since ubiquitination is involved in almost all biological processes, including protein homeostasis, transcription regulation, DNA repair, endocytosis and endolysosomal sorting, autophagy, immune response, and stem cell renewal, unsurprisingly, DUBs are also major players in these diverse cellular functions. Therefore, their dysregulation is associated with many diseases. This has been the subject of multiple recent reviews, either about their general functions [14][15][50][17,18,53] or on specific disease areas such as neuronal [51][54], cardiovascular [52][55], developmental [53][56], autoimmune [54][57] diseases, and cancers [55][56][58,59] and is beyond the scope of curthis rent contentview. Instead, these content we highlight the importance of using quality tool reagents when exploring the biology of DUBs at the cellular level. Due to the scarcity of quality DUB inhibitors, studies often use unvalidated, weak, or semi-selective inhibitors when probing for cellular functions. These practices can lead to spurious conclusions and add to the reproducibility crisis [57][58][60,61]. For example, compounds WP1130 and G9 have been used to examine the biological roles of USP9X, a large, 250-kDa USP family DUB. Studies using these compounds have claimed to elucidate numerous interactors and functions [59][62]; however, many of these interactors (for example, E3 ligases ITCH [60][63] and FBXW7 [61][64]; Halabelian and Tong unpublished data) and functions (Michael Clague personal communication) cannot be validated. One inhibitor, G9, which cross-reacts with USP5 and USP24 [62][65], has been used by many separate studies to assess the functions of USP9X [63][64][65][66,67,68], USP5 [66][67][69,70], or USP24 [68][71]. It is difficult to exclude the possibility of off-target effects, crosstalk, or functional compensation between these DUBs. On the other hand, FT709, a probe-quality inhibitor with a nanomolar affinity for USP9X, has provided much clearer evidence of its involvement in ribosomal stalling and confirmed its interaction with two other E3 ligases, ZNF598 and MKRN2 [69][72]. Another example is spautin-1 [70][73], which is widely used as a specific autophagy inhibitor for its inhibition of both USP10 and USP13. These two DUBs have very different structures. Whereas USP10 has a typical USP fold without insertions, USP13 has two ubiquitin-associated (UBA) domains inserted in the catalytic domain. USP13 is closer in sequence and domain architecture to USP5 than USP10. No orthogonal biophysical methods, such as the surface plasmon response (SPR) or isothermal titration calorimetry (ITC), were used to validate the binding of spautin-1 with USP10 or USP13.2. Small Molecule DUB Inhibitors
Despite a decade of intensive investment from industry and academic labs, most reported DUB inhibitors have a low binding affinity in the micromolar range and lack selectivity. This was demonstrated by a comprehensive profiling study using the MALDI-TOF mass spectrometry of 11 inhibitors against 42 DUBs [34][37] and by Medivir’s DUB platform [71][79]. ML323, an inhibitor of the USP1/UAF1 complex, was developed by Zhuang Lab [72][73][80,81] and stood to be the only USP family DUB inhibitor that met the chemical probe criteria [74][82] by 2016. However, the exact structural details on how ML323 binds to the USP1/UAF1 complex remained elusive, which prevented further structure-based optimization of the inhibitor. When several groups developed a series of high-quality, structurally defined USP7 inhibitors, the efforts in DUB inhibitor discovery were reinvigorated. The progress on USP7 inhibitors was nicely summarized in Pozhidaeva and Bezsonova’s review [75][83]. The demonstration of the druggability of USPs and the identification of structurally defined DUB inhibitors opened many opportunities to pharmacologically interfere with their functions in vivo and to explore their biology in disease states. The advances have been the subject of several recent excellent reviews [76][77][78][79][77,84,85,86] and will not be repeated here. In the last several years, the chemical biology community has been advocating a stringent evaluation of chemical tool reagents for the study of the biology of protein targets [76][80][81][76,77,78]. These criteria include <100 nM in vitro potency, >30-fold selectivity against other members of the same protein family, off-target profiling, and cellular on-target effects at <1 ¦ÌM. A combination of orthogonal validation methods to confirm target engagement is essential, especially considering the various shortcomings of some popular inhibitors (discussed above). HWe havinge kept track of the high-quality chemical inhibitors of DUBs using a publicly accessible repository called UbiHub [82][87]. So far, only four USPs (USP7, USP1, USP9X, and USP30); one UCH (UCHL1); and one JAMM family DUB (CSN5) have chemical probe-quality small molecule inhibitors (Table 1). However, it is noteworthy that some of these are covalent inhibitors, such as FT385 for USP30 [83][88] and IMP–1710 for UCHL1 [84][85][89,90], which both utilize a cyanopyrrolidine warhead for modifying the catalytic cysteine residue. Despite their selectivity over other DUBs, they may have off-target reactivity towards other unrelated enzymes, such as dehydrogenases, as demonstrated for the UCHL1 inhibitor MT16-205 [84][89]. Thus, extra care must be exercised when using these inhibitors to probe the biology of the intended DUB targets. The best practice for small molecule inhibitors in studying the biology of protein targets is well-established [86][91]. Readers are encouraged to follow the do’s and don’ts when using chemical probes to obtain credible results.| Characterization | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Target | IC50/Ki/Kd (nM) | MoA | Structure (pdb) | Negative Control | Validation | Ref. |
| IMP-1710 | UCHL1 | 38 | covalent, slowly reversible | IMP-1711 | cell-based assays; dose-response; in vivo target engagement; chemical proteomics | [85][90] | |
| CSN5i-3 | CSN5 | 5.8 | non-covalent | 5jog | R,R-CSN5i-2e | enzyme assays; cell-based assays; dose-response; XRD | [87][92] |
| Azaindole derivatives cmpds 4, 6 1 | CSN5 | 90, 60 | non-covalent | 5m5q 2 | enzyme assays; NMR; SPR; CellTiter-glo; XRD | [88][93] | |
| FT671 | USP7 | 52 | non-covalent | 5nge | mutagenesis; cell-based assays; SPR; MS | [89][94] | |
| GNE6640 | USP7 | 750 | non-covalent | 5uqv | GNE-6641 | enzyme assays; cell-based assays; NMR; MS; XRD | [37][40] |
| ALM-4, ALM-5 | USP7 | 1.5, 22 | non-covalent | 5n9t 3 | ent-ALM-4 | cell-based assays; SPR; target engagement assay; XRD | [90][95] |
| Pyrimidinone derivatives 4 | USP7 | 6−87 4 | non-covalent | 6f5h 5 | enzyme assays; cell-based assays; in vitro ADME assay; XRD | [91][96] | |
| XL177A | USP7 | 0.34 | covalent | XL177B | enzyme assays; cell-based assays; ABPP-MS; HDX | [92][97] | |
| XL188 | USP7 | 90 | non-covalent | 5vs6 | XL203C | enzyme assays; cell-based assays; ITC; XRD | [93][98] |
| FT709 | USP9X | 82 | non-covalent | cell-based assays; proteomics; SPR; ELISA | [69][72] | ||
| ML323 | USP1/UAF1 | 76 | non-covalent | NCGC-959 | enzyme assays; cell-based assays; orthogonal gel-based assays | [73][81] | |
| MF-094 | USP30 | 120 | non-covalent | MF-095 | enzyme assays; mitophagy assay | [94][99] | |
| FT385 | USP30 | 1 | covalent | enzyme assays; cell-based assays; BLI; ABPP | [83][88] | ||