Pharmacogenetics (PGx) is an emerging field of pharmacology focusing on how gene variations affect the patient’s response to treatment. Pharmacogenetics is a promising tool to optimize the selection and dosing of medications, including urate-lowering therapies (ULTs) among patients with gout. The global prevalence of gout is rising, and it disproportionately affects specific racial groups and individuals with select socioeconomic status. Genetic and experimental findings have provided evidence that genetic polymorphisms associated with serum urate pathology are also of pharmacogenetic interest. Patients with gout present with several comorbidities, warranting the use of several acute and long-term medications that increase their pill burden and the risk of adverse drug events. Implementing PGx testing can identify individuals who are more or less likely to benefit from a given treatment, improve medication adherence, and reduce pill burden.
- gout
- pharmacogenetics
- precision medicine
- genetics
- pharmacogenomics
- urate-lowering therapy
1. Background
2. Genetics of Hyperuricemia and Gout
Two-thirds of SUA is eliminated through the renal proximal tubule (RPT), while the remaining one-third is eliminated through the small intestine and metabolized by the gut microflora (Figure 1) [9][17].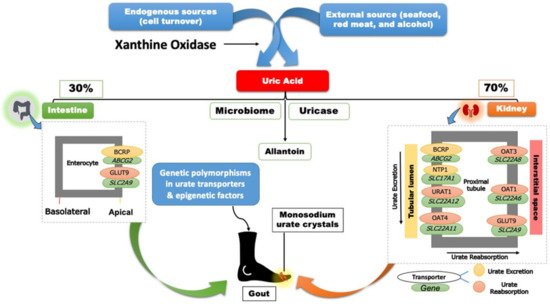
3. Gout Management Pharmacotherapy
The pharmacotherapy management of gout includes rapid and effective control of the inflammation in acute gout flares, continued ULT to prevent future flares, and ultimately improve gout treatment outcomes [2][5]. Contemporary gout treatment guidelines recommend allopurinol as the preferred first-line treatment for managing chronic gout. Pharmacotherapies, such as non-steroidal anti-inflammatory drugs (NSAIDs), colchicine, and corticosteroids, are also appropriate first-line agents to manage gout flares [2][5]. Pharmacotherapies, including interleukin-1 inhibitors (i.e., canakinumab and rilonacept), are also used to control gout flares when alternatives are contra-indicated or ineffective. Social and environmental factors, as well as diet and genetics, could affect a patient’s adherence and response to ULTs. As the field of pharmacogenomics continues to evolve, multiple studies have evaluated the effect of gene variants, including G6PD, HLA-B*58:01, and CYP2C9, on predicting the response to ULTs and possible adverse drug reactions (Table 1) [2][19][5,27].| Drug | Mapped Genes | Effect | Clinical Outcomes | CPIC Guideline Level of Evidence | a | References | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Xanthine oxidase inhibitors (XO) | ||||||||||
| Allopurinol or Oxypurinol | HLA-B | Safety | HLA-B*58:01 | allele significantly increases the risk of allopurinol-induced serious cutaneous reaction | A | [2][20] | [5,28] | |||
| AOX | Response | rs3731722 A> | G | is associated with a better response to the standard dose of allopurinol (300 mg/day) vs. non-carriers | NA | [21] | [29] | |||
| ABCG2 | Response/PK | rs2231142 C> | A | (Q141K) is associated with poor response to allopurinol | NA | [22] | [30] | |||
| SLC22A12 | Response/PK | rs505802 C> | T | may influence the response to allopurinol and the PK of oxypurinol as they are substrates for the URAT1 | NA | [23][24][25] | [11,31,32] | |||
| Febuxostat | UGT1A1 | Response/PK | rs34650714 C> | T | is associated with lower doses of febuxostat | NA | [21] | [29] | ||
| Uricosuric Agents | ||||||||||
| Probenecid | SLC22A12 | Response | Homozygous or heterozygous for the mutant allele (G774 | A | ) have impaired response to loading tests of probenecid | NA | [26][27] | [33,34] | ||
| ABCB1 | PK | rs1045642 C> | T | could influence the PK effect of probenecid as an inhibitor when co-administered with Beta-lactam | NA | [28] | [35] | |||
| G6PD | Safety | Possible hematologic adverse reactions in | G6PD | deficient patients | B | [29] | [36] | |||
| Benzbromarone | CYP2C9 | Safety | Carriers of the no-function allele ( | CYP2C9*3) | have reduced metabolic activity leading to prolonged exposure to benzbromarone relative to normal metabolizers | NA | [30][31] | [37,38] | ||
| Recombinant Uricase | ||||||||||
| Pegloticase | G6PD | Safety | Risk of hemolysis or methemoglobinemia in | G6PD | deficient patients | B | [32] | [39] | ||
| Non-steroidal anti-inflammatory drugs (NSAIDs) | ||||||||||
| Ibuprofen, celecoxib, and other NSAIDs | CYP2C9 | Safety/PK | Increased risk of NSAID-related GI bleeding in no-function allele ( | *3 | ) carriers relative to normal function, as well as reduced metabolism and prolonged exposure to ibuprofen and celecoxib in CYP2C9 poor metabolizers | A (ibuprofen and celecoxib); C (indomethacin, diclofenac, naproxen) | [33][34] | [40,41] | ||
| Anti-inflammatory | ||||||||||
| Colchicine | CYP2D6 | Response | Diminished response to colchicine in | CYP2D6*4 | variant carriers | NA | [35] | [42] | ||
| ABCB1 | Inconsistent evidence wherein one study indicates good response in the T allele carriers of the SNP rs10455642 C> | T | , while another study suggests no response with the T allele | NA | [36][37] | [43,44] | ||||
| SEPHS1 | Safety | The risk allele G of rs74795203 A> | G | significantly increases the risk of gastrointestinal adverse events by 2.5-fold with using colchicine | NA | [38] | [45] | |||
| KIF13A, RNU6-793Pb | Safety | The risk allele A of rs6916345 G> | A | (intergenic) was significantly associated with a ~2-fold increased risk of gastrointestinal adverse events with colchicine compared with the G allele | NA | [38] | [45] | |||
| Corticosteroids | ||||||||||
| Injectable triamcinolone acetonide | HCG22 | Safety | The G and T alleles of rs3873352 C> | G | and rs2523864 C> | T | , respectively, increase the risk of steroid-induced ocular hypertension | NA | [39] | [46] |
| IL-1 inhibitor | ||||||||||
| Anakinra | IL1RN | Response | SNP cluster in strong linkage disequilibrium associated with poor response to anakinra | NA | [40] | [47] | ||||