Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Joanna Zawitkowska and Version 3 by Catherine Yang.
Immunotherapy is a milestone in the treatment of poor-prognosis pediatric acute lymphoblastic leukemia (ALL) and is expected to improve treatment outcomes and reduce doses of conventional chemotherapy without compromising the effectiveness of the therapy. However, both chemotherapy and immunotherapy cause side effects, including neurological ones.
- acute lymphoblastic leukemia
- neurotoxicity
- immunotherapy
- chemotherapy
1. Introduction
Five-year overall survival of ALL has increased over the past decades and now exceeds over 96% [1]. Chemotherapy is a crucial part of treating ALL and involves many cytotoxic drugs, which inhibit cancer cells from growing rapidly, but they also damage healthy cells, resulting in a wide range of adverse effects. However, with the current high rate of survival, it would be difficult to improve results with only conventional chemotherapy which has reached its maximum of tolerance and could no longer be pushed to improved results. The target should be to search for the use of intensive multimodal treatment regimens, including high-dose chemotherapy and next-generation drugs [2]. Precision medicine with immunotherapy and other molecularly targeted treatments offers unique opportunities to customize treatment intensity [3]. Their advantages also include reducing the need for hematopoietic stem cell transplantation (HSCT), decreasing the burden of toxicities, and fighting persistent residual disease. Recently approved agents for ALL include blinatumomab, inotuzumab ozogamicin (InO), and CAR T-cell therapy, which are expected to improve treatment outcomes and reduce doses of conventional chemotherapy without compromising the effectiveness of the therapy. Nevertheless, the benefits of aggressive chemotherapy versus target therapy for different patient groups remain unclear and all the strategies cause adverse events, such as neurotoxicity, hepatotoxicity, gastrointestinal complication, and secondary malignancies, making neither of these therapies an ideal treatment [4].
2. Neurotoxicity of Conventional Therapy
The optimal treatment doses are determined based on tolerability, response assessment, and drug pharmacodynamics and pharmacogenomics. The clinical characteristics of the patient and the biological features of the leukemia are the main factors that determine the choice of specific chemotherapeutics. Treatment protocol consists of phases such as induction, intensification, consolidation, and maintenance [5][7]. Currently, first-line treatment protocols include a variety of medication combinations which involve the use of VCR, L-ASP, corticosteroids, antimetabolites (cytarabine and MTX), and anthracyclines [3]. However, the dose intensity of conventional chemotherapy has been pushed to its limits, and because children absorb and metabolize drugs differently than adults, toxicity is a key issue in pediatric chemotherapy. To determine further treatment development, attention must be given to some of the unique neurotoxicities associated with MTX, VCR, L-ASP, and their molecular background (Table 1).Table 1.
Treatments used in ALL and associated neurotoxicity.
| Phase of Treatment | Drugs | Toxicity-Related Gene | Mechanism of Neurotoxicity | Neurotoxicity | References | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Induction | Vincristine | ABCC11 | 1 | , | ABCC2 | 2 | , | ABCC4 | 3 | , | ABCC5 | 4 | , | ABCB1 | 5 | , | ABCC10 | 6 | , | CEP72 | 7 | , | SLC5A7 | 8 | , | TUBB1 | 9 | , | TUBB2A | 10 | , | TUBB2B | 11 | , | TUBB3 | 12 | , | TUBB4A | 13 | , | MAP4 | 14 | , | CYP3A4 | 15 | , | CYP2C8 | 16 | , | CYP3A5 | 17 | , | CEP72 | 18 | Interferes with the assembly of microtubule structures leading to cell apoptosis. It affects the peripheral nerves but can also contribute to dysfunction of the cranial nerves and autonomic nervous system. | Peripheral neuropathy, sensory neuropathy: symmetry sensory/tactile impairment, numbness, and tingling in the hands and feet, paresthesia, decreased balance, tendon weakening, visual and hearing problems. | [6][7] | [8,9] | ||||||||||||||||||
| L-asparaginase | ZBTB1 | 19 | , | GRIA1 | 20 | , | HLA-DRB1 | 21 | L-asparaginase produces three neurotoxic agents: ammonia, L-aspartic acid, and glutamic acid. These two amino acids can induce cell death in CNS neurons by excessive stimulation through NMDA (N-methyl-D-aspartate) receptor, leading to a major intracellular calcium influx and apoptosis. | Myelosuppression, encephalopathy, hepatic toxicity. | [8][9][10] | [10,11,12] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Consolidation | Methotrexate (intravenous infusion and intrathecally) | DHFR19bp | 22 | , | MTHFR 677C > T | 23 | , | MTHFR 677TT | 24 | , | SLC19A1 | 25 | , | TYMS | 26 | , | ADORA2A | 27 | Methotrexate is an antimetabolite that inhibits Dihydrofolate Reductase and thus tetrahydrofolate formation. This affects the synthesis of macromolecules such as myelin, and reversible leukoencephalopathy has been suggested to be secondary to impaired myelin turnover. Dihydrofolate Reductase inhibition leads to lack of folate and cobalamin, and increase in homocysteine, which is toxic to vascular endothelium may cause seizures and vascular disease. Dihydrofolate Reductase inhibition results in decreased levels of S-adenosylmethionine, which in turn plays a role in maintaining the myelin sheath, and this deficiency may lead to demyelination after intrathecal methotrexate administration. |
Transverse myelopathy-symptoms include back pain with subsequent weakness, sensory loss and bladder or bowel incontinence, blurred vision, aphasia, anarthria, seizures, aphasia, mental status disorder, stroke-like episodes, delirium, leukoencephalopathy septic meningitis characterized by headache, neck stiffness, nausea, vomiting and potential fever and encephalopathy. | [11][12][13] | [13,14,15] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cytarabine | DCK | 28 | , | NT5C2 | 29 | , | CDA | 30 | , | RRM1 | 31 | , | GIT1 | 32 | , | NT5C 3 | 33 | , | ENT1 | 34 | , | SCL29A1 | 25 | Cytarabine exhibits preferential toxicity for CNS | 35 | progenitor cells and oligodendrocytes, compromises cell division in vitro, and causes cell death and reduced cell division in vivo. | Myelosuppression, neurotoxicity. | [14][15][16][17] | [16,17,18,19] | |||||||||||||||||||||||||||||||||||||||||||||||
| Maintenance | Methotrexate (orally) | Genes have been described above. | Mechanism has been described above. | Seizures, aphasia, mental status disorder, stroke-like episodes, delirium, leukoencephalopathy, cognitive dysfunction, personality changes. | [11][12][13][18] | [13,14,15,20] |
1 ABCB11, ATP Binding Cassette Subfamily C Member 11; 2 ABCC2, ATP Binding Cassette Subfamily C Member 2; 3 ABCC4, ATP Binding Cassette Subfamily C Member 4; 4 ABCC5, ATP Binding Cassette Subfamily C Member 1; 5 ABCB1, ATP Binding Cassette Subfamily B Member 1; 6 ABCC10, ATP Binding Cassette Subfamily C Member 10; 7 CEP72, Centrosomal Protein 72; 8 SLC5A7, Solute Carrier Family 5 Member 7; 9 TUBB1, Tubulin Beta 1 Class VI; 10 TUBB2A, Tubulin Beta 2A Class IIa; 11 TUBB2B, Tubulin Beta 2B Class IIb; 12 TUBB3, Tubulin Beta 3 Class III; 13 TUBB4A, Tubulin Beta 4A Class Iva; 14 MAP4, Microtubule-Associated Protein 4; 15 CYP3A4, Cytochrome P450 Family 3 Subfamily A Member 4; 16 CYP2C8, Cytochrome P450 Family 2 Subfamily C Member 8; 17 CYP3A5, Cytochrome P450 Family 3 Subfamily A Member 5; 18 CEP72, Centrosomal Protein 72; 19 ZBTB1, Zinc Finger and BTB Domain Containing 1; 20 GRIA1, Glutamate Ionotropic Receptor AMPA Type Subunit 1; 21 HLA-DRB1, Major Histocompatibility Complex Class II, DR Beta1; 22 DHFR19bp, Dihydrofolate Reductase 19 bp polymorphism; 23 MTHFR 677C > T, Methylenetetrahydrofolate Reductase polymorphism; 24 MTHFR 677TT, Methylenetetrahydrofolate Reductase polymorphism; 25 SCL29A1, Solute Carrier Family 29, member 1; 26 TYMS, Thymidylate Synthetase; 27 ADORA2A, Adenosine A2a Receptor; 28 DCK, Deoxycytidine Kinase; 29 NT5C2, 5’-Nucleotidase, Cytosolic II; 30 CDA, Cytidine Deaminase; 31 RRM1, Ribonucleotide Reductase Catalytic Subunit M1; 32 GIT1, G Protein-Coupled Receptor Kinase Interacting ArfGAP 1; 33 NT5C3, 5’-Nucleotidase, Cytosolic IIIA; 34 ENT1, Equilibrative nucleoside transporter 1; 35 CNS, central nervous system.
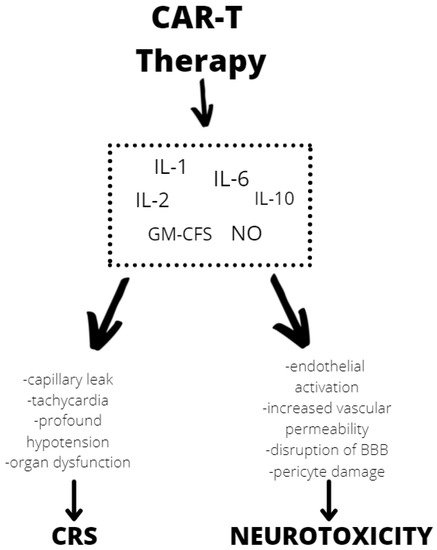
 Although the exact mechanism of neurotoxicity is unknown, evidence shows endothelial activation and increased blood–brain barrier permeability, which results in a high cytokine concentration in the cerebrospinal fluid. These cause more endothelial cell and pericyte activation, which, if severe, might result in cerebral edema or other CAR T neurotoxicity manifestations (Figure 2). In contrast to the previously assumed mediators derived from T lymphocytes, after CAR T infusion thwe reseachers observed a significant increase in the level of cytokines, including granulocyte-macrophage colony growth factor (GM-CSF), IL-10, IL-6, and IL-1 and IL-6 generated from host macrophages that have been shown to mediate neurotoxicity. Depending on the kinetics of T-cell proliferation, CRS with CAR T might develop shortly after infusion or be a delayed response that occurs days or weeks later [34][81].
Although the exact mechanism of neurotoxicity is unknown, evidence shows endothelial activation and increased blood–brain barrier permeability, which results in a high cytokine concentration in the cerebrospinal fluid. These cause more endothelial cell and pericyte activation, which, if severe, might result in cerebral edema or other CAR T neurotoxicity manifestations (Figure 2). In contrast to the previously assumed mediators derived from T lymphocytes, after CAR T infusion thwe reseachers observed a significant increase in the level of cytokines, including granulocyte-macrophage colony growth factor (GM-CSF), IL-10, IL-6, and IL-1 and IL-6 generated from host macrophages that have been shown to mediate neurotoxicity. Depending on the kinetics of T-cell proliferation, CRS with CAR T might develop shortly after infusion or be a delayed response that occurs days or weeks later [34][81].
 The evaluation and classification of these toxicities vary greatly between clinical trials and institutions, making it difficult to compare the safety of different medicines and to create effective care methods for these toxicities. The first patients treated with CD19 CAR T-cells experienced similar toxicities to those observed with the immunomodulatory drug theralizumab (TGN1412), including aggressive behavior, rigidity, fever, poor concentration, and psychosis [35][82]. Supraphysiologic cytokine increase was found to be responsible for the great majority of symptoms in the first pediatric ALL patient treated with CAR T-cell therapy, implying that these toxicities were caused by CRS. Encephalopathy, agitation, aphasia, tremor, lethargy, delirium, difficulty concentrating, seizures, and in rare cases even cerebral edema are all symptoms of ICANS [36][83]. In addition, headache is a very common symptom that may or may not indicate neurotoxicity (Figure 3). Over the years, many scales have been developed, such as CTCAE (Common Terminology Criteria for Adverse Events) version 4.03, CTCAE version 5.0, Lee criteria, and Peen criteria et al., which redefined the scoring criteria for CRS. Many CAR T-cell groups have adopted the Lee criterion, in part because it was the first to link a specific grade to a suggested therapy protocol. As previously indicated, neurotoxicity is a common side effect of CAR T-cell and other T-cell-engaging therapies. Neurotoxicity associated with immune effector cells, unlike classic CRS symptoms, do not often respond to tocilizumab treatment. This fact is not surprising, given that when tocilizumab is administered I.V., large amounts of the drug do not accumulate in the CSF. Symptoms of ICANS can be more diverse than those of CRS. Many patients with neurotoxicity have a stereotypic evolution of a specific set of symptoms. The earliest manifestations of ICANS are mild difficulty with expressive speech (especially in naming objects), dysgraphia, impaired attention, tremor, apraxia, and mild lethargy [37][84]. Early detection of marker changes such as peak C-reactive protein (CRP), ferritin on day 3 but not peak ferritin, and fever can have a huge impact on the prognosis of patients who develop neurotoxicity. In people at risk of neurotoxicity, the number of cytokines such as IL-6, IL-10, granulocyte-macrophage colony stimulating factor, IL-15, IL-2, and the TNF receptor should also be determined. However, it is important to realize that none of these markers are unique to chemotherapy or immunotherapy or CRS induced neurotoxicity. Only the combination of several ingredients gives a picture of whether a given patient is at risk of neurotoxicity. High-dose methylprednisolone is typically used in the most severe cases of CAR T-cell-related neurotoxicity. Most neurotoxic patients also have CRS, which can be treated with tocilizumab, an IL-6 receptor antibody, and/or corticosteroids to suppress T-cell activation, but the effect of these medications on neurotoxicity is uncertain [38][85].
The evaluation and classification of these toxicities vary greatly between clinical trials and institutions, making it difficult to compare the safety of different medicines and to create effective care methods for these toxicities. The first patients treated with CD19 CAR T-cells experienced similar toxicities to those observed with the immunomodulatory drug theralizumab (TGN1412), including aggressive behavior, rigidity, fever, poor concentration, and psychosis [35][82]. Supraphysiologic cytokine increase was found to be responsible for the great majority of symptoms in the first pediatric ALL patient treated with CAR T-cell therapy, implying that these toxicities were caused by CRS. Encephalopathy, agitation, aphasia, tremor, lethargy, delirium, difficulty concentrating, seizures, and in rare cases even cerebral edema are all symptoms of ICANS [36][83]. In addition, headache is a very common symptom that may or may not indicate neurotoxicity (Figure 3). Over the years, many scales have been developed, such as CTCAE (Common Terminology Criteria for Adverse Events) version 4.03, CTCAE version 5.0, Lee criteria, and Peen criteria et al., which redefined the scoring criteria for CRS. Many CAR T-cell groups have adopted the Lee criterion, in part because it was the first to link a specific grade to a suggested therapy protocol. As previously indicated, neurotoxicity is a common side effect of CAR T-cell and other T-cell-engaging therapies. Neurotoxicity associated with immune effector cells, unlike classic CRS symptoms, do not often respond to tocilizumab treatment. This fact is not surprising, given that when tocilizumab is administered I.V., large amounts of the drug do not accumulate in the CSF. Symptoms of ICANS can be more diverse than those of CRS. Many patients with neurotoxicity have a stereotypic evolution of a specific set of symptoms. The earliest manifestations of ICANS are mild difficulty with expressive speech (especially in naming objects), dysgraphia, impaired attention, tremor, apraxia, and mild lethargy [37][84]. Early detection of marker changes such as peak C-reactive protein (CRP), ferritin on day 3 but not peak ferritin, and fever can have a huge impact on the prognosis of patients who develop neurotoxicity. In people at risk of neurotoxicity, the number of cytokines such as IL-6, IL-10, granulocyte-macrophage colony stimulating factor, IL-15, IL-2, and the TNF receptor should also be determined. However, it is important to realize that none of these markers are unique to chemotherapy or immunotherapy or CRS induced neurotoxicity. Only the combination of several ingredients gives a picture of whether a given patient is at risk of neurotoxicity. High-dose methylprednisolone is typically used in the most severe cases of CAR T-cell-related neurotoxicity. Most neurotoxic patients also have CRS, which can be treated with tocilizumab, an IL-6 receptor antibody, and/or corticosteroids to suppress T-cell activation, but the effect of these medications on neurotoxicity is uncertain [38][85].

3. Neurotoxicity of Immunotherapy
Several of the newly discovered molecular alterations have led to the development of approaches that focus on the dysregulation of cellular pathways [19][5]. Despite tremendous advances in the treatment of ALL, no drug has shown more promise in improving survival outcomes than immunotherapeutic approaches. However, increasing evidence suggests that immunotherapy also induces serious and complex neurologically related adverse events and may even lead to related deaths, raising concern among clinicians for their more widespread use [20][53]. Nevertheless, neurotoxicity caused by immunotherapy should not be underestimated. Early and accurate diagnosis of neurotoxicity increases the effectiveness of treatment, but its mechanism of neurotoxicity has not yet been fully elucidated [21]. For this reason, treatment of neurotoxicity is limited only to the clinician’s own experience.3.1. Blinatumomab
Dual-specific T-cell-binding antibodies (BiTEs) are made up of two antibodies with varying antigen-binding domains joined by a non-immunogenic binding peptide. One of the most clinically advanced BiTEs is blinatumomab, generated from a B-lineage-specific mouse monoclonal antibody. Blinatumomab is made up of two arms: one for attaching CD3-expressing T-cells and another for attaching CD19+ B-cells. Blinatumomab is approved for the treatment of precursor B-ALL in first or second complete remission with minimal residual disease (MRD) greater than or equal to 0.1% in adults and children [22][62]. There are reports that overall neurologic toxicities with blinatumomab treatment occur in 15–50% of patients. Neurologic adverse events can be severe, life threatening, or fatal, and grade 3 or higher neuropsychiatric toxicity has occurred in approximately 15% of patients [23][63]. The mechanism of blinatumomab-induced neurotoxicity is still unknown. However, CNS events are presumed to be caused by an inflammatory irritation of the myoendothelium by blinatumomab-activated T-cells, which locally release neurotoxic cytokines and chemokines locally on their way into the CNS [24][64]. Adverse neurologic events may include tremor, slurred speech, loss of vibratory sensation, dizziness, confusion, encephalopathy, and seizure, which are the most common manifestations of neurotoxicity. In adult patients, symptoms of neurotoxicity began after an average of 9 days, but pediatric patients may manifest earlier [25][65]. For patients with bone marrow blasts of more than 50%, peripheral blood blasts of 15,000 cells per μL or higher, or elevated lactate dehydrogenase suggesting rapidly progressing disease, some scientists required prephase treatment with dexamethasone 10–24 mg/m2 per day (for up to 5 days) to reduce the incidence of severe cytokine release syndrome. However, after the completion of the studies, it was found that pre-treatment with dexamethasone had no effect on the response [23][63].3.2. Inotuzumab Ozogamicin
Inotuzumab ozogamicin (InO) is an antibody–drug conjugate composed of a monoclonal CD22-directed antibody linked to calicheamicin, a potent cytotoxic antitumor antibiotic that induces apoptosis by breaking double-strand DNA [26][70]. In 2017, the FDA approved InO for treating adults with CD22-positive R/R B-cell precursor ALL [27][71]. Unfortunately, because of the small number of randomized trials of InO in children, it is difficult to estimate the incidence of neurotoxicity. There are no studies on the mechanism of neurotoxicity either [28][72]. A recent report from the COG study aimed to prospectively determine safety and efficacy of InO in pediatric and adolescent patients with R/R B-ALL. Patients in the InO arm received 1.8 mg/m2 intravenously each cycle, for a maximum of six cycles. Patients in the chemotherapy arm received cytarabine with mitoxantrone, FLAG (fludarabine, cytarabine, and granulocyte colony-stimulating factor [GCSF]), or high-dose cytarabine, as determined by the investigator. Among 48 patients, 19 patients achieved a complete response (CR), and 9 patients achieved a CR with incomplete cell count recovery (CRi) after cycle 1 of InO. However, in all studies using InO, hepatotoxicity, particularly sinusoidal obstruction syndrome (SOS), is one of the most life-threatening adverse events due to mortality rates exceeding 80% in patients who develop multiple organ failure. Side effects described in InO drug information related to CNS include only headache, which means that InO appears to be safe and well tolerated in terms of neurotoxicity [29][73]. Kantarjian et al. conducted a trial on 326 patients treated for ALL which were randomly assigned to receive either InO or standard intensive chemotherapy. The percentage of patients who had serious adverse events was similar in the InO group and the standard-therapy group: 48% and 46%, respectively. However, in neither group was death associated with neurotoxicity [30][74].3.3. CAR T-Cell Therapy
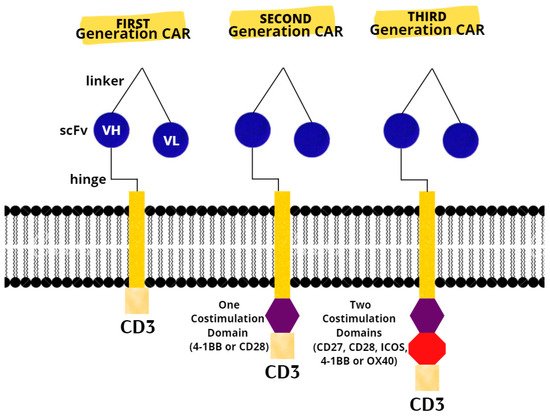
The last resort for acute or refractory ALL in patients up to 25 years of age are two CAR T products (tisagenlecleucel and axicabatagene ciloleucel) that have recently been approved for treatment in the United States and Europe [31][75]. There are three generations of CARs (Figure 1). A CAR T-cell’s basic structure usually consists of a tumor-targeting domain derived from a monoclonal antibody linked to a CD3 zeta chain that serves as an intracellular signaling domain. A co-stimulatory endodomain, either 4-1BB or CD28, is also present in second-generation CARs [32][76]. A third-generation CAR T-cell’s purpose is to increase T-cell proliferation and persistence by combining signaling domains such as 4–1BB, OX40 (CD134), inducible T-cell costimulator (ICOS), and CD27, to boost the cytotoxic effect [33][77].Figure 1. Chimeric antigen receptors. Next-generation CARs have additional modifications to their intracellular stimulatory domains. CD3, cluster of differentiation 3; ICOS, Inducible T-cell costimulator; scFv, single-chain fragment variable; VH, heavy chain variable gene segment; VL, variable region.
Figure 2. Mechanisms of neurotoxicity and cytokine release syndrome (CRS) caused by CAR T therapy. BBB—blood–brain barrier, GM-CFS—granulocyte-macrophage colony stimulating factor, NO—nitric oxide.
Figure 3. The management of ICANS is based on a grading system. CRS—cytokine release syndrome, ICANS—immune effector cell-associated neurotoxicity syndrome, ICE—Immune Effector Cell-Associated Encephalopathy score.