Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sandrine Dubrac and Version 2 by Rita Xu.
The discovery in 2006 that loss-of-function mutations in the filaggrin gene (
FLG
) cause ichthyosis vulgaris and can predispose to atopic dermatitis (AD) galvanized the dermatology research community and shed new light on a skin protein that was first identified in 1981. However, although outstanding work has uncovered several key functions of filaggrin in epidermal homeostasis, a comprehensive understanding of how filaggrin deficiency contributes to AD is still incomplete, including details of the upstream factors that lead to the reduced amounts of filaggrin, regardless of genotype.
- atopic dermatitis
- filaggrin
- skin
- microbiome
1. Filaggrin: A Look in the Rear-View Mirror
Filaggrin was initially isolated from a protein fraction of the stratum corneum (SC) and identified as a basic histidine-rich protein [1]. Proteins from this fraction were shown to aggregate with keratin filaments to form macrofibrils in in vitro cell-free experiments [2]. In 1981, Dale et al. designated this class of cationic structural proteins, which associate specifically with intermediate filaments but not with other types of cytoskeletal proteins, as filaggrins (for filament aggregating proteins) [2]. They showed that filaggrins are species-distinct proteins; for example, rat and mouse filaggrins have different molecular weights (48 and 30 kDa, respectively) and different amino acid totals but nevertheless exhibit similar chemical and functional properties. The ability of filaggrin to aggregate keratin filaments into tight parallel arrays [3] has been demonstrated in many different experimental settings, with high reproducibility [4][5][6][7][4,5,6,7]. This molecular bundling confers mechanical resilience and flexibility to the SC [8]. Mouse and human filaggrin bind to each three-chain building block of the intermediate filaments, possibly through ionic interactions with the coiled-coil alpha-helical regions of the keratin filaments. The stoichiometry of the interaction has been reported as two filaggrin molecules to three intermediate (keratin) filament subunits [3][9][3,9].
Further work demonstrated that filaggrin is produced as a phosphorylated precursor, profilaggrin, of 300–500 kDa, depending on the species [10][11][12][10,11,12], embedded in keratohyalin granules in granular keratinocytes (KCs) [2][13][14][2,13,14]. Subsequently, profilaggrin undergoes a multiple-step dephosphorylation process, followed by proteolytic cleavage of short linker peptides to produce various numbers of filaggrin monomers depending on the species, e.g., 10 to 12 in humans, 16 to 20 in mice [15][16][17][15,16,17], and only 4 in dogs [18]. Profilaggrin is present in the stratum granulosum (SG), whereas filaggrin monomers are localized in the first layers of the SC. Of note, there is often confusion between profilaggrin and filaggrin, with some authors incorrectly assigning filaggrin to the SG. Filaggrin, together with other proteins of keratohyalin granules, forms the protein moiety of the cornified cell envelope of corneocytes [19][20][21][22][23][19,20,21,22,23]. In rat corneocytes, filaggrin was found to represent about 10% of the cell envelope-bound proteins [24]. Early work suggested the involvement of one or more specific phosphatases in filaggrin dephosphorylation [11][25][11,25], and in 1988, a rat 40 kDa phosphatase specific for profilaggrin was isolated [26] and later characterized as a protein phosphatase 2A-type [25]. However, data concerning specific profilaggrin phosphatases in humans are scant.
In 1993, calpain 1 was proposed as a profilaggrin protease because profilaggrin cleavage requires calcium, and calpains are calcium-dependent neutral proteases [27]. In 2009, processing of human profilaggrin by human calpain 1 was confirmed in a cell-free proteolytic assay [28]. Moreover, cathepsin L-like proteinase, but not cathepsin E, isolated from rat epidermis was shown to hydrolyze profilaggrin [29]. Work in mice showed that loss of cathepsin H, produced by an shRNA approach, leads to a dramatic reduction of filaggrin monomers, owing to defective profilaggrin processing in the epidermis [30]. The results for cathepsin D are contradictory, and the enzyme is likely not essential for profilaggrin processing in vivo [29][31][32][29,31,32]. Furthermore, profilaggrin endoproteinase 1 (PEP1) has been identified as a cytoplasmic enzyme that is able to digest insoluble profilaggrin purified from mouse epidermis, at least in proteolytic assays [33]. In addition, both furin and PACE4, two calcium-dependent serine proteases belonging to the same family of proprotein convertases, can cleave profilaggrin in vitro at a site between the amino terminus and the first filaggrin repeat [34]. In mice, genetic deletion of matriptase/MT-SP1, a type II transmembrane serine protease expressed in epithelial cells, including KCs, leads to failure to process profilaggrin into filaggrin monomers [35]. Moreover, it has been shown that skin-specific retroviral-like aspartic protease (SASPase) activity is indispensable for processing profilaggrin in mouse epidermis [36], a requirement which may extend to human epidermis [37][38][37,38]. SASPase cleaves the linker sequence of human profilaggrin between ‘GSFLY’ and ‘QVSTH’ [36]. Moreover, it has been proposed that lympho-epithelial Kazal type inhibitor (LEKTI), a protease inhibitor encoded by the SPINK5 gene, controls profilaggrin proteolysis. Indeed, SPINK5 knockout mice or mice with a premature stop codon in SPINK5—both are mouse models of Netherton syndrome, a severe ichthyosis due to null mutations in SPINK5—display increased amounts of mature filaggrin and reduced amounts of profilaggrin, indicating that proteolytic processing of profilaggrin is enhanced [39][40][39,40]. In human KCs, it was further shown that mesotrypsin liberates a 55-kDa N-terminal fragment of profilaggrin, hence potentially contributing to filaggrin processing as well [41]. Lastly, in transgenic mice overexpressing elastase 2, the rate of profilaggrin processing into filaggrin monomers is accelerated. In line with this, in the SG of patients with Netherton syndrome, elastase 2 is upregulated and co-localizes with profilaggrin, and profilaggrin levels are reduced [42][43][44][42,43,44]. Thus, many different proteases have been shown to potentially cleave profilaggrin into filaggrin monomers.
In the upper SC, filaggrin is further processed by various proteases into free amino acids. The proteolysis of filaggrin is enhanced by its prior deamination or by citrullination. The latter is a post-translational modification catalyzed by peptidylarginine deiminase enzymes, resulting in the conversion of arginine into citrulline, thereby reducing its charge (citrulline is a neutral residue, whereas arginine is positively charged) and promoting detachment of the filaggrin monomer from the aggregated keratins [45][46][47][45,46,47]. Histidine and glutamine are then either enzymatically or spontaneously transformed to trans-urocanic acid (UCA) and pyrrolidone carboxylic acid (PCA), respectively. These amino acids and derivatives are components of the natural moisturizing factor (NMF), together with other molecules, such as lactate, chloride and sodium ions, and urea, thus ensuring proper SC hydration [48]. Interestingly, studies with histidinemic or caspase-14-deficient mice or with epidermal equivalents knocked-down for FLG have shown a role for trans-UCA in the protection of skin against the damaging effects of UV-B radiation [5][49][50][51][52][53][54][5,49,50,51,52,53,54].
In 1986, Scott and Harding established that proteolysis of filaggrin in rat skin is controlled by atmospheric humidity. Filaggrin is first detected at day 20 of rat gestation in all layers of the neo-formed SC. A few hours after birth, filaggrin disappears from the upper SC layers and accumulates in the innermost half of the SC, before being confined to a thin layer at the bottom of the SC, two days after birth. In addition, keeping the newborn rat in 100% humidity or applying occlusive patches onto the skin of adult rats prevents the activation of filaggrin degradation. The authors concluded that the lowering of atmospheric humidity promotes filaggrin proteolysis into water-retaining molecules [55]. This was confirmed first in mice [56] and then in humans, using 3D epidermal equivalents [57]. Indeed, when human epidermis is produced ex vivo at low atmospheric humidity, as compared to the typical saturated conditions, profilaggrin synthesis is increased, as is filaggrin degradation into UCA and PCA. This is in line with measurements conducted in humans, showing that NMF levels are low at birth and increase within hours or days after birth in healthy baby skin [58]. These results suggest that changes in the skin microenvironment, as occurs at birth, activate filaggrin degradation but not necessarily via increased protease activity. Indeed, it has been shown in vitro that reducing the external humidity increases filaggrin deimination and, as a consequence, the molecule’s dissociation from the filamentous corneocyte matrix [46][47][57][59][46,47,57,59]. This may render filaggrin more accessible to proteases [48]. Moreover, these results suggest that improper bundling of filaggrin with keratin filaments might promote filaggrin degradation in the absence of other alterations, such as increased activity of proteases. Besides external humidity, the neutral cysteine protease bleomycin hydrolase has been shown to participate in the breakdown of deiminated filaggrin into amino acids in a cell-free proteolytic assay [28]. Moreover, bleomycin hydrolase-deficient mice display reduced amounts of skin NMF and higher levels of filaggrin [60]. Furthermore, in healthy baby skin, regional upregulation of bleomycin hydrolase activity, e.g., in the cheek when compared to the elbow flexure, might contribute to increased skin hydration at specific body sites [58].
Thus, SASPase and calpain 1 might play important roles in the processing of profilaggrin into filaggrin, whereas bleomycin hydrolase and caspase 14 are probably major proteinases responsible for filaggrin breakdown in the skin (Table 1, Figure 1); however, the contribution of other proteinases cannot be ruled out.
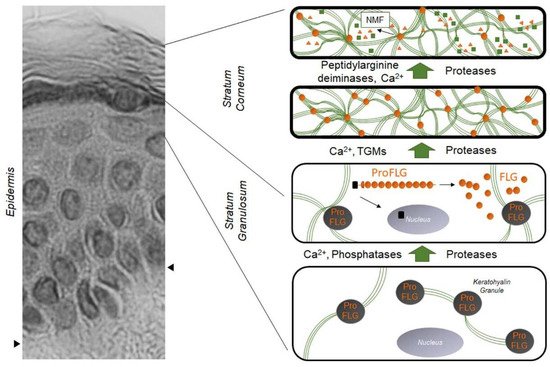
Figure 1. Evolution and distribution of profilaggrin and filaggrin during epidermal differentiation: dark gray disks containing profilaggrin are keratohyalin granules present in the cytoplasm of granular KCs. Under the action of phosphatases, proteases, and Ca2+, profilaggrin is expelled in the cytoplasm and degraded into filaggrin monomers (small orange disks). Green lines are keratin filaments aggregated by filaggrin monomers in the lower SC. Under the action of transglutaminases (TGMs), filaggrin molecules may be covalently linked to the cornified envelope. In the upper SC, filaggrin is further processed into free amino acids by the sequential action of peptidylarginine deaminases and proteases, producing the Natural Moisturizing Factor (NMF, green squares and orange triangles). The left part of the figure is a hematoxylin–eosin staining of a healthy human epidermis; arrows show the dermis–epidermis junction.
Table 1. Summary of enzymes and enzyme inhibitors involved in filaggrin metabolism (sometimes still hypothetic) in the epidermis according to data generated in vitro and in animal models versus in humans.
| In Vitro and Animal Models | in Humans | |
|---|---|---|
| Dephosphorylation of profilaggrin | Phosphatase of the protein phosphatase 2A family |
|
| Profilaggrin processing | Calpain 1, cathepsins L-like and H, PEP1, furin, PACE4, MT-SP1, mesotrypsin, SASPase, LEKTI, elastase 2 | Calpain I, SASPase, mesotrypsin |
| Filaggrin citrullination in the stratum corneum | PAD1 and/or 3 | PAD1 and/or 3 |
| Proteolysis of filaggrin in the stratum corneum | Bleomycin hydrolase, calpain 1, caspase 14 | Bleomycin hydrolase, caspase 14 |
2. The Impact of the FLG Gene: From Ichthyosis Vulgaris to Atopic Dermatitis
Ichthyosis vulgaris (IV) is a common skin disorder in humans, characterized by dry, rough, and scaly skin. In 1985, it was first reported that keratohyalin granules, profilaggrin, and filaggrin are all reduced or absent in the epidermis of patients with IV and that these biochemical abnormalities correlate with the clinical severity of the disease [61]. In 1991, high-resolution ultrastructural immunolabeling showed many small filaggrin-positive granules in association with bundles of keratin filaments in IV SG [20]. However, unequivocally, the amounts of filaggrin are much lower in the SC of IV patients compared to that of healthy volunteers [20]. In 2006, genetic studies shed new light on the IV disorder, with the identification of loss-of-function mutations in the FLG gene associated with moderate or severe IV [62]. In the same year, Palmer et al. showed that FLG loss-of-function mutations are strongly associated with atopic dermatitis (AD) [63], suggesting that low amounts of filaggrin in AD might result from genetic causes. Indeed, earlier pioneering reports showed a decrease of filaggrin in lesional AD epidermis [64], regardless of skin lesion presence or absence [65][66][65,66]. AD, also known as atopic eczema, is the most common inflammatory skin diseases, affecting 1%–36% of children and up to 18% of adults, of whom approximately 20% have moderate-to-severe disease [67][68][69][70][67,68,69,70]. Here it is important to emphasize that reduced filaggrin levels are also observed in AD patients who are genotypically the wild type for FLG [65][71][65,71], demonstrating that FLG variants are not the only factors responsible for filaggrin downregulation in AD skin, and that other factors (e.g., environmental, metabolic) are also likely to be involved. Moreover, African American patients with AD exhibit normal levels of filaggrin, in contrast to Asian and European patients [66][71][66,71]. On the other hand, pediatric AD, regardless of the patient’s ethnic background, might be characterized by normal skin levels of filaggrin [72]; however, most studies lack either FLG genotyping or measurements of filaggrin amounts [64][72][73][74][64,72,73,74].
The half-life of rodent filaggrin in the SC is 6–9 h, demonstrating that filaggrin degradation is a relatively rapid process [16][75][16,75]. This high turnover of filaggrin may confer vulnerability to the epidermis because factors that lead to reduced filaggrin expression or that accelerate its degradation could have rapid effects on the biological processes in which filaggrin is involved. Conversely, environmental factors or therapies able to augment filaggrin expression might have rapid beneficial effects. Thus, it is important to decipher accurately all the physiological roles of filaggrin in the epidermis so that pathways potentially ameliorable by filaggrin-targeted therapy are fully identified.
3. FLG Null Mutations Are Strong Genetic Factors in AD
The strong association between FLG null mutations and AD (OR = 13.4), first observed by Palmer et al. [63], was subsequently confirmed by many other research groups in studies of various ethnic populations [76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102]. The strength of this association was initially reported to depend on FLG genotype; that is, in a cohort of 186 patients with AD, homozygosis for the combined null genotype of six major null mutations (R501X, 2282del4, R2447X, S3247X, 3702delG, and 3673delC) was more strongly associated with the disease (OR = 85.9) compared to heterozygosis (OR = 4.6) [103]. Furthermore, Brown et al. found, in a large cohort of children, that heterozygosis is not significantly associated with AD (OR = 1.2), as opposed to homozygosis (OR = 26.9) [104]. Nomura and Kabashima suggest that the initial overestimation of the strength of the association of FLG null mutations with AD, especially in heterozygous patients, is likely due to recruitment of patients with severe disease symptoms [67]. Nevertheless, it remains that FLG homozygosis or compound heterozygosis significantly increases the risk of developing AD, at least in adult European and Asian populations [71][105][71,105]. However, Morar et al. found FLG null mutations in 26.7% of young patients with AD, but also in 14.4% of children without AD [106]. Subsequent studies have confirmed that about 40%–50% of all carriers of FLG null alleles never experience eczema [107][108][107,108]. Thus, these data show strong but incomplete penetrance of FLG variants [63][80][109][110][111][63,80,109,110,111], whose impact on AD development might be modulated by ethnic-specific genetic modifiers, epigenetic alterations, or other environmental factors.
Beyond FLG mutations, the number of filaggrin monomers encoded by an FLG allele can vary from 10 to 12 in humans. Individuals with 20 copies of the filaggrin monomer (i.e., homozygous 10/10) were reported to have an increased risk of developing AD, whereas those with 21 to 24 copies (genotypes 10/11 to 12/12) had no significantly elevated risk, at least in patients with moderate AD [112]. Similar work in patients with low-to-mild AD would give additional information on the power of the association between the different numbers of filaggrin monomers (i.e., genotypes) and the disease expression.
FLG null mutations have been shown to correlate with AD severity and persistence in adulthood [77][88][106][113][114][77,88,106,113,114]. Indeed, the observed higher prevalence of adult AD patients with an FLG null mutation (42%) [113] when compared to pediatric patients (13%) [104] argues for a role of FLG status in disease evolution, although this remains to be confirmed in large patient cohorts with different ethnic backgrounds.
A new classification of AD patients according to age, ethnicity, concomitant atopy, inflammatory milieu, SC lipid abnormalities, and FLG status designated as ‘endotypes’ has recently been proposed. In this perspective, AD patients with FLG null mutations represent a particular endotype [66][115][66,115].