Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Anna Lierová and Version 2 by Conner Chen.
Hyaluronic acid (also known as sodium hyaluronate or hyaluronan) is a straight-chain, natural polysaccharide and the only nonsulfated GAG composed of alternating (1–4)-β d-glucuronic and (1–3)-β N-acetyl-d-glucosamine units [5]. Both carbohydrate units are spatially related to glucose; therefore, in the β-configuration, it is possible for all their bulky groups (hydroxyl and carboxyl groups and the anomeric carbon on the neighboring sugar) to be in sterically favorable planes, while all the small hydrogen atoms occupy less sterically favorable axial positions. This chemical structure of HA is energetically very stable because of interactions between hydrophobic and intermolecular hydrogen bonds and the acetamide and carboxylate groups.
- hyaluronic acid
- synthesis
- degradation
1. Introducing Hyaluronic Acid
The importance of hyaluronic acid (HA) has increased over the past 10 years due to new biomedical applications exploiting its full biocompatibility and unique bioreactivity. HA is a polysaccharide belonging to the group of glycosaminoglycans (GAGs), which is a group of highly sulfonated, complex, linear polysaccharides manifesting a number of important biological roles. HA is in several respects an exception among GAGs. Within the cell, GAGs are synthesized in the Golgi apparatus network and subsequently bind covalently to proteins while proteoglycans are being created. Based on their distinct, repeating disaccharide units, GAGs can be divided into four major groups: heparin/heparan sulfate, chondroitin sulfate/dermatan sulfate, keratan sulfate, and hyaluronan. These polymers serve as cell surface molecules or in extracellular matrix (ECM) [1][2][3][4][1,2,3,4].
Hyaluronic acid (also known as sodium hyaluronate or hyaluronan) is a straight-chain, natural polysaccharide and the only nonsulfated GAG composed of alternating (1–4)-β d-glucuronic and (1–3)-β N-acetyl-d-glucosamine units [5]. Both carbohydrate units are spatially related to glucose; therefore, in the β-configuration, it is possible for all their bulky groups (hydroxyl and carboxyl groups and the anomeric carbon on the neighboring sugar) to be in sterically favorable planes, while all the small hydrogen atoms occupy less sterically favorable axial positions [6]. This chemical structure of HA (Figure 1) is energetically very stable because of interactions between hydrophobic and intermolecular hydrogen bonds and the acetamide and carboxylate groups [7].
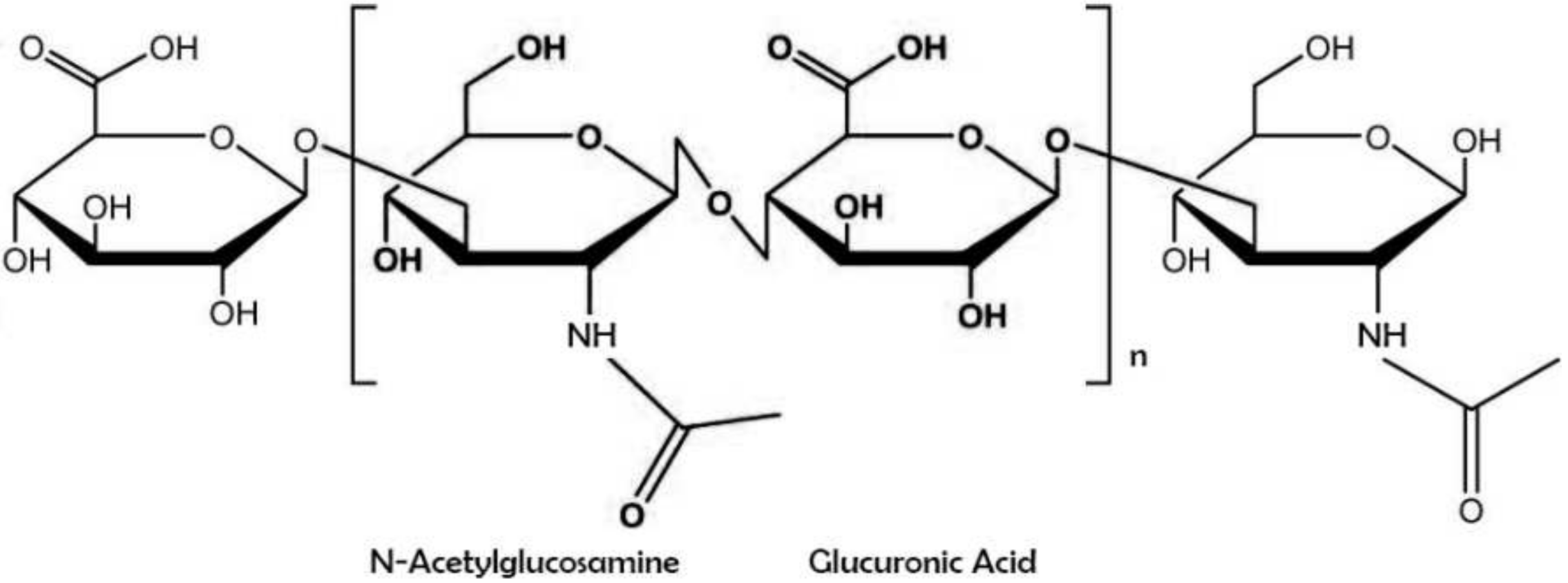
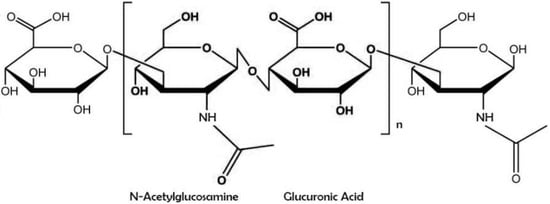
Figure 1. Chemical structure of hyaluronic acid with repeating disaccharide units of (β, 1–4)-glucuronic acid (GlcUA) and (β, 1–3)-N-acetyl glucosamine (GlcNAc).
HA was first isolated from the bovine eye vitreous body by Karl Meyer and John Palmer in 1934 [8]. The name “hyaluronic acid” was created as a conjugation of the two words: hyaloid (vitreous body) and uronic acid. Although Meyer and Palmer are generally considered to be the discoverers of HA, the very first mention of the HA molecule dates back to 1918, when Levene and Lopez-Suarez isolated an unknown polysaccharide from a vitreous body and umbilical cord blood that they called mucoitin sulfuric acid [9]. The precise chemical structure of HA was described after almost 20 years of research also by Meyer’s group [10].
Hyaluronan is one of the most hygroscopic molecules found in nature [11]. The molecular chains are intertwined to form highly viscous and elastic solutions, even at very low HA concentrations. This phenomenon can be observed in solutions containing as little as 1 μg/mL HA, which is one of the reasons for this molecule’s unique rheological properties [12]. The strong hydrophilicity of HA is, therefore, the physical basis for its widespread presence in the body. HA is found in the ECM of all vertebrates’ tissues [13], but it is also as a component of an extracellular capsule produced naturally by various species of bacteria [14]. In the human body and especially in connective and soft tissues, HA exists primarily in the form of high molecular weight chains (HMW-HA) of greater than 10 kDa [15]. HA hydrophilicity is the fundamental and a very important property for the control of tissue hydration and osmotic balance [16]. Due to its strong anionic nature, HA can capture water molecules with winding chains, leading to an important physicochemical property: water retention. The large number of hydroxyl groups greatly contributes to this property [17]. HA can retain water up to 1000 times its own weight [18]. The water content increases with rising relative humidity [19]. Hydration parameters are, nevertheless, independent of HA’s molecular weight [20].
One of the most controversial capabilities of hyaluronan in the body is its antioxidant activity as an effective “scavenger” of free radicals. It is well known that reactive oxygen species (ROS) can lead to oxidative stress, which implies a change in intracellular redox homeostasis and leads to a serious imbalance between the production of reactive species and antioxidant defense [21]. On the one hand, ROS degrades HA [22]; on the other, the antioxidant capacity of hyaluronic acid is scientifically proven [23]. Many years of intensive research and numerous published scientific papers have been needed to clarify this incongruity. More precise mechanisms will be explained in the next section, because this feature is critical for the intention of our review.
2. A Unique Metabolism from Beginning to End
HA is uniquely synthesized on the inner side of the plasma membrane by membrane-bound glycosyltransferases. From a molecular biology perspective, these HA glycosyltransferases disproved the molecular dogma that “one enzyme carries only one sugar group” [24][26]. The key year for this discovery was 1993, when DeAngelis et al. for the first time successfully identified, sequenced, and cloned hyaluronan synthase A (hasA), a gene from Streptococcus pyogenes [25][26][27,28]. A second turning point came in 1997, when a second group of HAS genes was identified in bacteria Pasteurella multocida (PmHAS), also by a team led by DeAngelis [27][29]. Although this is the only PmHAS of the latter group known to date, chondroitin synthase proteins that are structurally similar have been identified and cloned in two bacteria: P. multocida and Escherichia coli [28][30]. Since that time, more than 20 eukaryotic, bacterial, and even viral genes or cDNA sequences have been described for HAS [28][30]. Based on the more advanced technology and methods used in genome identification and sequencing, whether prokaryotic or eukaryotic, and the amount of experimental results obtained, some authors have already suggested that it would be appropriate to classify HAS into three groups [29][31]. It is beyond the scope of theise contents review to explain or even mention all the basic information concerning the individual species of HAS, but this topic is described in great detail in several excellent reviews (such as DeAngelis [24][26], Weigel [30][32], and Siiskonen et al. [31][33]). Within the scope of our article, we will only briefly mention HAS in mammalian species, basic information, and topics that are currently of great and particular interest.
In vertebrae species, three isoforms of HA synthase (HAS1–3) have been identified (except that the genus of Xenopus laevis has 4 genes [32][34]) that are specifically expressed in various time intervals and tissues under different physiological or pathological conditions [33][35]. These are evolutionarily quite highly conserved proteins, sharing 50–71% of amino acid sequences, but the gene sequence of each isoform is localized on a different chromosome (HAS1—hCh19; HAS2—hCh8; HAS3—hCh16) [34][35][36,37]. In general, HAS1 synthesizes HMW-HA with masses 2 × 105 to 2 × 106 Da and produces chains of variable lengths, but is the least active isoform. In contrast, HAS3 synthesizes mainly HA polymers of low molecular weight (LMW-HA; 1 × 105 to 1 × 106 Da), but it is the most active of all isoforms. Chains produced by the HAS2 isoform are also high molecular weight HA, similarly as are those of HAS1, but HAS2 tends to produce more uniform chains with masses greater than 2 × 106 Da [36][37][38][38,39,40]. In contrast with the physiological properties of individual HAS enzymes, in the work of Itan et al., with recombinant proteins of individual mammalian HAS enzymes, the highest Michaelis constant (KM) value was reached by HAS1, which may cause the lower synthetic rate of nucleotides [39][41]. This study also suggests that the concentrations of individual nucleotide precursors, as well as the overall nature and intrinsic properties of individual synthases, are important factors in the regulation of HA chain creation. One of the most critical discoveries at the turn of the millennium in hyaluronan science was clarification as to the roles of these synthases in the stages of embryogenesis and, particularly, those of HAS2. Camenisch et al. found that HAS-deficient mice embryos (HAS2−/−) have severely impaired cardiac and vascular morphogenesis and develop highly severe or lethal defects [40][42]. Nevertheless, HAS1−/− and HAS3−/− mice, as well as double knockouts, are viable and fertile [41][43]. Of particular importance is mainly HAS2, which is the most abundant isoform in adult tissues and is involved in the processes of tissue development, repair, growth, and regeneration in the cases of insult. For these reasons, the expression and impact of the HAS2 isoform also will be important to the aims of our review and will be discussed further in the next section.
Each HAS enzyme is capable of de novo chain synthesis (Figure 2), and the differences are the molecular weights of the chains being created and, hence, in their biological functions [42][44]. HAS uses cytosolic UDP-N-acetylglucosamine and UDP-glucuronic acids (UDP-GlcNAc and UDP-GlcUA) as precursors to polymerize the HA chain without the need for a primer, anchor protein, or lipid [43][45]. The amounts of nucleotide precursors and their relative proportions constitute one of the major limiting factors of the HA chain, and the results from mass spectrometry analyses confirm that HAS1 produces also chitin oligomers in the presence of UDP-GlcNAc and absence of UDP-GlcUA [44][46]. The number of repeating disaccharides on a completed hyaluronan molecule may reach 10,000 or more disaccharide units, and the molecular weight can grow to as great as 4 × 106 Da. The average length of a disaccharide is approximately 1 nm. Thus, a hyaluronan molecule of 10,000 repeating units could elongate by 10 μm, which is approximately the diameter of a human erythrocyte [45][47]. After synthesis, HA is extruded into extracellular space without any need for transport or exocytosis.
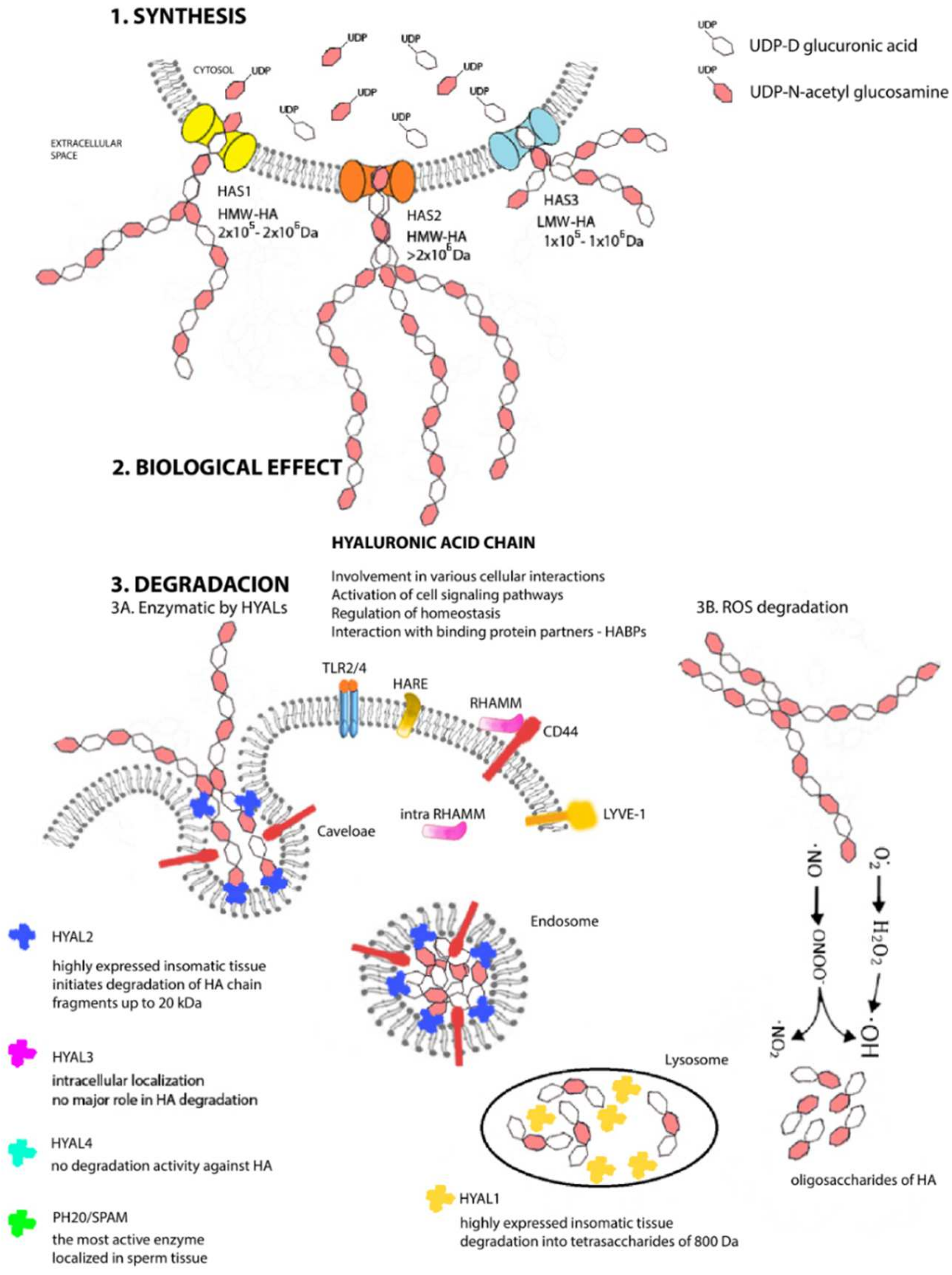
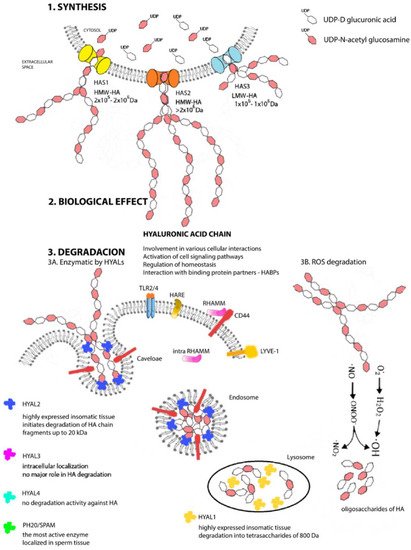
Figure 2. Metabolism of hyaluronan. Structural scheme of synthesis (1), biological effect (2), and degradation (3) of HA under physiological conditions in mammal cells. Cytosolic molecules of UDP-D glucuronic acid and UDP-N-acetyl glucosamine serve as precursors for HA chains. The enzymatic reaction is catalyzed by three HAS proteins, which synthetize unique HA chains varied in lengths that differ in their biological function in the organism (1). Hyaluronic acid chains are involved in many cellular interactions, in signaling pathways, binding activities with other proteins, or are involved in process of homeostasis (2). In vivo degradation proceeds in parallel in two ways (enzymatically (3A) and chemically (3B)). Specific hyaluronidases (HYAL1—HYAL3 and PH20/SPAM) are localized in different tissue of organism and initiates the degradation of HA chain (HYAL1 in lysosomes, HYAL2 in inner part of endosome membranes, and HYAL3 in cellular membrane). Enzyme PH20 SPAM is localized in testis. Free radicals provide the random chemical degradation (3B) of HA chains and after a longer exposure, fragmentations occur with a consequent increase in the number of small HA oligosaccharides.
Intensive research on HA-related enzymatic structures is still ongoing today because the precise mechanisms of their translocation to cell membrane, the regulation of the molecular transcription mechanism and post-transcriptional modifications, and even the complete physiological roles of individual HAS enzymes are not yet known [46][48]. The expression of Has genes is also significantly influenced by the increased activity of growth factors and cytokines or signaling molecules under certain conditions in tissues [47][49]. The work of Tlapak-Simmons et al. was the first to unambiguously determine the direction of HA synthesis in individual groups by confirming that class I HAS enzymes polymerize chains at the reducing end [48][50]. The current capabilities for co-immunoprecipitation and Förster resonance energy transfer (FRET) analysis are rewriting and adding to the knowledge about HAS functions and actions. Deen et al. identified the Rab10 protein as the first suppressor for trafficking to the plasma membrane, which is responsible for controlling HAS3 levels [49][51]. Karousou and her research team confirmed that HAS2 is regulatory ubiquitinated [50][52], which affects activity and stability and further demonstrates that HAS2 is capable of dimerization. The work of Bart et al., however, has shown that all HAS isoenzymes form homomeric and heteromeric complexes with one another. They even have suggested that the homomeric and heteromeric interactions detected among the different HAS isoenzymes are specific and potentially influence the enzymatic activity of native HAS proteins, perhaps in a dominant negative manner [51][53].
HA is a ubiquitous component of the ECM and pericellular space, evolutionarily occurring already from bacteria, algae, and mollusks; some viruses; and to mammalians from among chordates, including humans [52][54]. It has not yet been observed, however, in fungi, plants, or insects (although insect venom contains enzymes cleaving HA (hyaluronidases) [53][54][55,56]. The fruit fly Drosophila melanogaster, however, can produce HA in vivo after transfection with the mouse HAS2 gene, albeit with significant morphological defects due to the bulk accumulation of HA in the extracellular space [55][57]. In an adult human weighing 70 kg, the amount of HA present is approximately 15 g. More than one-half of this amount is present in the skin (primarily in the intercellular space of the dermis), in the synovial fluid of the knee joint there is approximately 2–3 mg/mL, and the vitreous body of the eye contains approximately 200 µg/mL [56][57][58,59]. Moreover, the umbilical cord contains a significant amount of HMW-HA (up to 4 mg/mL) [10]. HA is nevertheless present in all body tissues and fluids, for instance, in lungs (15–150 mg/g), where it is mainly localized in the peribronchial and interalveolar space [58][60], in skeletal muscles [59][61], as well as in lymph. Surprisingly, a lymph has been found to contain primarily LMW-HA and serves as its pool [60][62].
In the body, HA occurs in many different forms, including circulating [61][63], bound by various HA-binding proteins (known as hyaladherins) [62][64], associated with tissue or ECM [63][65], or electrostatically or covalently bound to other matrix molecules [64][66]. HA does not bind directly by covalent, chemical bonds to the protein core; therefore, it does not form proteoglycans [65][67], but it is known that it can form aggregates with proteoglycans occurring in ECM [66][67][68,69].
Moreover, the degradation of HA is permanent, leading to extremely rapid turnover of HA in the body. HA’s transformation times range from a few minutes (in the bloodstream) to 2 days, but a maximum of up to 70 days in the vitreous of the eye [68][70]. Degradation takes place at different locations, with approximately 30% being degraded locally in tissues and the remaining 70% entering the lymphatic drainage [69][71]. Degradation in vivo can take place via two main and simultaneous mechanisms: enzymatic degradation and chemical depolymerization [70][72]. The enzymatic degradation of HA occurs via the endoglycosidase family of enzymes, consisting of homologous proteins generally referred to as hyaluronidases (HYALs) and specifically hydrolyzing the β-1,4 linkage predominantly in HA molecules [71][73]. Recently, it has been confirmed that HYALs have limited ability to slowly degrade other GAGs, namely chondroitin and chondroitin sulfates [72][74]. This group of enzymes was also classified by Meyer in 1971 [73][75]. Based upon this exceptionally precise classification, HYALs are divided into three classes with different cleavage end products: bacterial lyases (EC 4.2.99.1) [74][76]; endo-β-glucuronidases occurring in leeches and bark beetles (EC 3.2.1.36) [75][77]; and those exceptionally cleaving the β-1,3 bond and mammalian hydrolases (EC 3.2.1.35) [76][77][78,79]. In humans, six gene sequences for HYALs have been identified to date: hyaluronidases 1–3 (HYAL1–3) are localized on hCh3p21.3, while HYAL4, PH20/SPAM, and HYALP1 pseudogenes are clustered on hCh7q31.3 [71][73]. The expression of individual HYALs is tissue-specific [78][80]. We will Tbriefly mention, at least, the basic information about the individual HYALs will be briefly mentioned.
The first to be isolated and purified was HYAL1 [79][81], a 57-kDa polypeptide glycoprotein that is the major degradative enzyme present in serum [80][82], a cell cycle regulator, and a candidate tumor suppressor gene [81][83]. HYAL2 is a glycosylphosphatidylinositol (GPI)-linked enzyme with low activity attached to the outside of the plasma membrane; it cooperates with HA receptors present on the cell surface and primarily with a cluster of differentiation 44 (CD44) [82][84]. Both HYAL1 and HYAL2 are highly expressed in human somatic tissues and are optimally active at acidic pH [83][85]. HYAL1 works together and sequentially with HYAL2 to degrade HA. HYAL2 initiates the degradation of HMW-HA chain into fragments of up to 20 kDa (approximately 50–60 disaccharide units), which are transported first to endosomes and then to lysosomes [84][86], where HYAL1 steps in and by its action, they are degraded into tetrasaccharides of 800 Da, a predominant end-product of HYALs degradation [85][87]. The resulting oligosaccharides become substrates for the terminal reaction of two lysosomal β-exoglycosidases: for D-glucuronidase, which hydrolyzes terminal nonreducing glucuronic acid, and for p-N-acetyl-D-hexosaminidase, hydrolyzing nonreducing UDP-GlcNAc [76][78]. The catabolic pathway of HA generates different size fragments, which widely differ in their biological HA properties. HA catabolism is a highly ordered, carefully controlled process under the regulation of the individual enzyme activities. Due to that, the LMW-HA chains are very active inflammatory biological molecules; actually, tetrasaccharides produced by HYAL1 can be recognized as “dangerous”. It also has been found that hyaluronan depositions and turnovers are even more intense and rapid in patients’ tumor tissues with a larger proportion of LMW-HA [86][88]. Biological properties of different HA chain are described in next section, but due to this biological divergence, a precise control mechanism must exist. The inhibitors of HYALs are ubiquitously present in every tissue, but this class of molecules is still not fully known, and their mechanism remains undiscovered [87][88][89,90].
Interestingly, HYAL4 and PH20/SPAM also are GPI-anchored proteins. PH20/SPAM1 is the most active, multifunctional GPI-linked sperm protein important for fertilization. It is localized on the anterior surface of sperm that facilitates the surrounding the oocyte. It also degrades HA chains from oligo- to tetrasaccharides. Testicular tissue is a physiological source of bioactive hyaluronidase [89][91]. Although currently available evidence indicates that HYAL4 has no degradation activity against HA, it predominantly and uniquely degrades chondroitin sulfate [90][92]. The latest discovery is hyaluronidase protein Transmembrane Protein 2 (TMEM2) located on the cell’s surface and is strongly active in the pH of the extracellular environment. This protein does not share structural homology with other HYALs, but it has 48% amino acid homology with cell migration-inducing proteins (CEMIP; discussed below). Neither the identification of TMEM2 nor its role in HA catabolism has been completely resolved [91][93]. Not much is known, either, about the activity and functions of individual HYALs. Such information as is currently available and only fuels more new questions and the need for intensive research. How is it possible that HYAL1 is the only protein present in the circulation and at the same time a degradative lysosomal enzyme in a pH 4.5 environment? HYAL3 also is a mystery because, despite its widespread expression in various tissues but very low activity, it cannot be identified by available hyaluronidase assays [92][94]. The clarification of these questions will lead to a more detailed understanding of the degradation pathway but also of the overall HA metabolism and signaling pathway, implying new, more advanced modulation strategies in situations of tissue damage or tumor disease.
The second method of degrading HA, chemical depolymerization, is intermediated through reaction with ROS, including superoxide, hydrogen peroxide, nitric oxide, hypohalous acids, and hydroxyl and peroxynitrite radicals at the glycosidic bond [93][95]. The result is the subsequent fragmentation of the polymer into various LMW-HA chains up to the size of oligomers. Free radicals and HYALs may coordinate their activities in certain pathological situations, but the relative roles of the different mechanisms are not known [94][96]. The depolymerization of HA will be discussed in detail in the next section, because this is a direct consequence of IR on the body and a product of this reaction is the significant disturbance of the equilibrium at the level of physical, chemical, and especially biological homoeostasis of the extracellular matrix. With its versatile properties, such as its biocompatibility, non-immunogenicity, biodegradability, and viscoelasticity, HA is an ideal biomaterial finding wide application, particularly in the cosmetic industry and in non-surgical aesthetic dermatology [95][97] and ophthalmology [96][98]. In the new millennium, it also has become a very important molecule in tissue engineering [97][99], drug-delivery systems [98][100], and cancer therapy [99][101].