Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ilya Ulasov and Version 2 by Rita Xu.
Multiple efforts are currently underway to develop targeted therapeutic deliveries to the site of glioblastoma progression. The use of carriers represents advancement in the delivery of various therapeutic agents as a new approach in neuro-oncology. Mesenchymal stem cells (MSCs) and neural stem cells (NSCs) are used because of their capability in migrating and delivering therapeutic payloads to tumors.
- stem cells
- glioblastoma
1. Introduction
Glioblastoma (GBM) is the most common and aggressive primary tumor in adults. According to the classification of the World Health Organization (WHO), glioblastoma belongs to the fourth type of malignancy of intracranial neoplasms. Even though this type of tumor is the most common representative of primary brain tumors, the incidence of the disease in humans is relatively low—3.1 cases per 100,000 population (for comparison, 171.2 and 201.4 cases per 100,000 population were noted for breast and prostate tumors, respectively) [1]. It is well-established that the uncontrolled growth of glial cells stimulates aggressive tumor growth that may lead to the death of patients. Anticancer therapy could increase the survival of patients with neoplasms. A group of scientists demonstrated that the survival rate within five years does not exceed 5%, and the median survival rate reaches 15 months after treatment, including surgery combined with radiation and chemotherapy. As shown earlier [2], the low survival rate of patients with GBM is based on the adaptive response of tumor cells to therapy. An adaptive response is expressed in the activation of the growth of tumor cells by an autocrine mechanism and the induction of cellular factors that cause inflammation and immunosuppression of the tumor [3][4][5][3,4,5]. The standard methods of treatment do not increase the survival rate. Experimental methods of glioblastoma therapy are being actively developed that reach the effectiveness of the existing ones. On the other hand [6], the use of cancer vaccines, which are based on the activation of the acquired immunity of a patient in response to tumor-specific antigens [7], is promising for experimental oncology. A vaccine can be both individual peptides [8][9][8,9], which is promising for experimental oncology, and whole autologous antigen-presenting cells [10]. However, the use of a platform based on human and animal viruses seems to be a more effective approach to inducing an immune response in tumors. This is explained by the fact that the virus can be used to deliver and express a library of tumor antigens and succinates the activation of the immune response and the elimination of tumors and metastases. On the other hand, the natural mechanism of viral replication makes it possible to obtain recombinant proteins in significant volumes. There are approaches involving the use of viruses, both as vectors for gene therapy [11] and as oncolytic agents [12], delivering immunogenic proteins or antisense RNAs to tumor proteins that promote carcinogenesis [13]. At the same time, native immune responses can be one of the obstacles that decrease the anticancer efficacy of viral vectors against cancer cells [6]. For example, the pool of the neutralization of antibodies against human adenovirus type 5 decreases the efficacy of CRAd (Conditionally replicated adenoviruses)-based vectors using the human adenovirus type 5 genome [14][15][14,15]. Additionally, low diffusion through the tumor mass [16] and the activation of cellular protein kinases [17], for example, modulate the inflammatory immune response and overall decrease the therapeutic response to the selected vectors. To overcome those issues and still specifically deliver a gene therapy vector to the target cells, stem cell carriers can be of interest.
2. General Characteristics of Mesenchymal Stem Cells (MSC)
Mesenchymal stem cells (MSCs) are a heterogeneous population of fibroblast-like cells. MSCs represent a small population (0.01% of all nucleated cells found in bone marrow) [18][22]. MSCs are multipotent and differentiate into chondrocytes, skeletal myocytes, and neurons, if applicable. As noted earlier, more than 95% of MSCs express a high level of CD73, CD90, and CD105 and a low level of protein II (the major histocompatibility complex (MHC-II)) and CD45, CD34, CD14, CD11b, and CD19 on their surface. MSCs can express other markers in various combinations dependent on the tissue specificity. In 2011, Ode et al. provided a mechanistic insight into the biological significance of surface receptors for MSC migration. CD73/CD29 were among the antigens that were described in the study and implemented in MSC migration. It turns out that MSC migration is controlled by CD73/CD29 and their downstream targets such as Lck, Fyn, and Src-family kinase activation [19][23].3. MSC—Cell Vectors for Migration and Delivery of Therapeutic Proteins
MSCs expressing protein molecules have been used in preclinical studies to successfully deliver antigens and induce a therapeutic effect in human and animal glioblastoma cells. As previously stated, stem cells were used to deliver viruses [20][21][22][24,25,26], suicide proteins from herpes simplex viruses like thymidine kinase (TK) [23][24][25][27,28,29], cellular miRNA [26][30], exosomes [27][31], apoptosis-induced proteins like TNF (tumor necrosis factor)-related apoptosis-inducing ligand (TRAIL) [28][32], cytosine deaminase (CD) [29][33], carboxylesterase 2 (sCE2, a prodrug-activating enzyme) [30][34], antiangiogenic thrombospondin-1 [31][35], and chemical agents such as paclitaxel [32][36] that activates the T-cell response in glioblastomas [33][37] (Figure 1).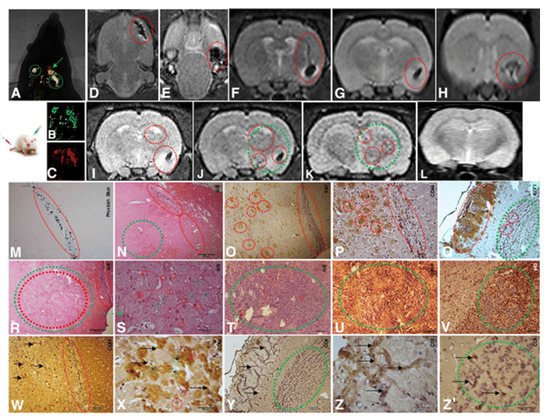
Figure 1. Implantation of 1 × 104 CD133+ GBM cells labeled Qdots (705 nm). After the establishment of the GBM (28 days), there was an infusion in caudal vein 1 × 104 MSCs (MION-Rh); the development of the tumor was followed for 20 days. (A) MSCs labeled MION-Rh and CD133+ GBM cells labeled Qdot 705 nm using combined fluorescence and X-ray detection. (B) CD133+ GBM cells labeled Qdot 705 nm and visualized by fluorescence detection. (C) MSCs labeled MION-Rh and visualized by fluorescence detection. (D–L) MRI (T2*-weighted images) of animal brain monitoring of the process of migration of MSCs, which were able to cross the blood–brain barrier of the animal and migrated to the tumor region, promoting GBM cell proliferation. (L) MRI (T2*-weighted images) of the animal brain without the stereotaxic implantation of cells (control group). The red dot circle shows migration assays of MSCs, and the green dot circle shows tumor propagation. (M) IHC analysis for Prussian blue staining of the MSCs labeled with MION-Rh. (N,R–T) Hematoxylin and eosin staining. (U) IHC analysis for GFAP. (O) IHC analysis for Ki67. (V) IHC analysis for p53. (P) IHC analysis for CD44 staining of the MSCs. (Q) IHC analysis for CD73 staining of the MSCs. (W,X) IHC analysis for CD63 staining of the MSC-derived exosomes. (Y,Z,Z′) IHC analysis for CD9 staining of the MSCs-derived exosomes. Scale, 40 µm. These images are representative of all the collected MSCs and GBM samples. Image and text were copied from the original study by Pavon et al. [34][42] and distributed under Attribution 4.0 International (CC BY 4.0).
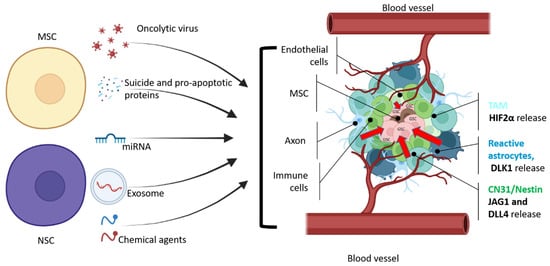
Figure 2. General scheme of therapeutic agents that can be delivered by NSC or MSC cells to glioma stem cells. GSCs are located in the necrotic area of the tumor, which is surrounded by hypoxic and vascularized regions with leaky blood vessels (adapted from Vinogradov et al. [52][56]). CD31+/Nestin+ and reactive astrocytes produce JAG1, DLL4 [53][57], and DLK1 [54][58] to perinecrotic and perivascular tumor regions to maintain GSC stemness and proliferation. According to Kvisten et al., [55][59] tumor-associated astrocyte (TMA) areas are rare in necrotic areas and more distributed in perivascular and perinecrotic areas of the GBM. Despite the morphological and phenotypical differences, TMA produces HIF-2α [56][57][60,61], which also controls the GBM pathobiology. In order to deliver a therapeutic payload, NSCs or MSCs should overcome multiple obstacles that prevent GSCs from being eliminated. Cell microenvironment releases growth factors to maintain GSC (red arrows).