Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Zaneta Kimber-Trojnar and Version 3 by Jason Zhu.
Endometriosis is a common disease in women of reproductive age, and its pathogenesis seems to be largely affected by hormone imbalance, inflammation, oxidative stress, and autophagy dysregulation. Metformin is an insulin sensitizer widely used for the treatment of type 2 diabetes mellitus. In endometriosis, metformin might modify the stroma–epithelium communication via Wnt2/β-catenin. With its unique therapeutic mechanisms and no serious side effects, metformin seems to be a helpful anti-inflammatory and anti-proliferative agent in the treatment of endometriosis.
- Metformin
- Endometriosis
- Mechanisms
1. Metformin
Metformin is an antidiabetic medicament currently used as the first-choice treatment for type 2 diabetes mellitus (T2DM), which does not cause excessive hypoglycaemia [1][59]. Metformin leads to a significant reduction in plasma fasting insulin levels and a reduction in insulin resistance. Therefore, it is considered an insulin sensitizer, which seems to be associated with its beneficial effects on tyrosine kinase activity and insulin receptor expression [2][60]. Metformin, by regulation of multiple components of the incretin axis, may have a positive effect on metabolism. Maida et al. [3][61] presented that this agent stimulates gene expression of islet incretin receptor by a mechanism that is dependent on peroxisome proliferator-activated receptor (PPAR)-α and intensely raises levels of glucagon-like peptide 1 (GLP-1) in plasma.
Nevertheless, a great deal of argumentation from clinical researchers confirms that the fundamental action of metformin is to reduce intrahepatic glucose production, mostly by inhibiting gluconeogenesis through a mild and temporary inhibition of the mitochondrial respiratory chain complex 1 [4][5][6][62,63,64]. Furthermore, the decline in hepatic energy status initiates the AMP-activated protein kinase (AMPK), which provides a mechanism for metformin function on the hepatic gluconeogenic program.
Metformin also decreases obesity-associated inflammation and other inflammatory reactions and affects the steroidogenesis in ovarian granulosa and thecal cells [7][8][9][10][11][12][13][65,66,67,68,69,70,71]. Metformin inhibits plasminogen activator inhibitor-1 (PAI-1) levels, endometrial androgens receptor expression, and plasmatic endothelin-I (ET-1), which are factors that increase the risk of miscarriage.
The therapeutic potential of this medicament, which is most likely induced by the improvement in insulin sensitivity, is employed in different conditions, including cardiovascular diseases, diabetic nephropathy, polycystic ovary syndrome (PCOS), and the prevention or treatment of cancer [1][14][15][59,72,73] (Figure 1). Hyperinsulinemia caused by insulin resistance might, in fact, induce carcinogenesis indirectly by raising the levels of steroid sex hormones and directly through the insulin receptor or insulin-like growth factors (IGF), inflammatory processes, and interrupting adipokines homeostasis [16][74].
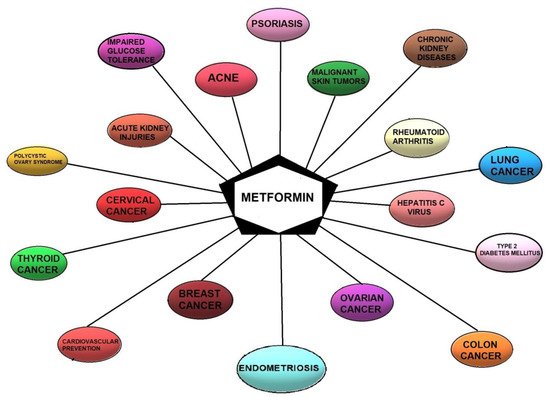
Figure 1. Current and potential therapeutic indications for metformin.
2. Metformin as a Potential Treatment Option for Endometriosis—Mechanisms
Metformin has a pleiotropic effect by stimulating AMPK, which is the major regulator of cellular energy homeostasis that inhibits mTOR, the main suppressor of autophagy [17][18][19][55,75,76]. The activation of AMPK is of particular importance for understanding the mechanism of metformin’s action in women with endometriosis. The AMPK, as a serine/threonine-protein kinase, is a principal enzyme that regulates energy homeostasis in cells [7][65]. Furthermore, mitogen-activated protein kinase (MEK)/extracellular-signal -regulated kinases (ERK) phosphorylation is triggered by metformin [20][21][77,78].
Zhou et al. [7][65] reported that the mechanism of metformin action is the inhibition of prostaglandin E2 (PGE2)-induced cytochrome P450 19A1 (CYP19A1) mRNA expression and aromatase activity in endometriotic stromal cells (ESC) by inhibiting the binding of the cAMP-Response Element Binding protein (CREB) to the Proximal Promoter (PII). This process was associated with the activation of AMPK. It should be emphasized that profuse CYP19A1 mRNA expression and elevated local oestrogen production have been found in endometriotic tissues, implying that P450 aromatase is involved in the local oestrogen production [7][65]. Changes in the expression of CYP19A1 mRNA may result in a significant decrease in aromatase activity.
Additionally, it has been confirmed that metformin inhibits the mRNA expression of insulin-stimulated CYP19A1 and follicle-stimulating hormone (FSH) in granulosa cells [10][22][68,79] and also significantly decreases the forskolin/phorbol ester (FSK/PMA) -dependent upregulation of CYP19A1 mRNA expression in primary stromal cells of human breast adipose tissue [23][80]. The intracellular actions related to these various effects of metformin in ESCs rely on the fact that the AMPK pathway is the main mechanism of metformin action [7][23][24][25][26][27][28][65,80,81,82,83,84,85]. This means that metformin can reduce CYP19A1 mRNA expression and aromatase activity to some extent independently of AMPK [7][65].
Nevertheless, metformin regulates the expression of aromatase using other signalling pathways and unknown mechanisms. Furthermore, the AMPK pathway is gradually phosphorylated upon metformin stimulation in ESC [20][77]. A similar effect on the gene expression of CYP19A1 in human ESCs might have the AMPK inhibitor and PGE2 [7][65]. In addition, adiponectin, whose concentrations are reduced in serum and peritoneal fluid in endometriotic patients, stimulates AMPK and inhibits the production of inflammatory cytokines in endometriosis [29][30][31][86,87,88]. Moreover, AMPK participates in the anti-inflammatory action of metformin shown by ESCs [21][78]. These observations confirm that induction of AMPK in endometriosis may be a protective factor. Steroidogenesis and CYP19A1 gene expression have been shown to be regulated by the MEK/ERK signalling cascade; however, conflicting findings regarding the mechanism in various steroidogenic cells have been reported. For instance, it has been reported that inhibition of MEK activity with PD98059 and U0126 is associated with stimulation, inhibition, or no effect on the steroidogenic response [20][21][32][33][34][35][77,78,89,90,91,92]. Zhou et al. [7][65] demonstrated the inhibition of CYP19A1 mRNA expression by metformin as autonomous of the MEK/ERK pathway.
Stimulation of the MEK/ERK pathway may be relevant in other metformin mechanisms. Metformin may reduce PGE2-activated CREB binding to CYP19A1 PII in human ESCs [7][65]. Notably, metformin decreases CYP19A1 expression at the transcriptional level. It is noteworthy that metformin interacts with the FSH-stimulated cAMP/PKA/CREB pathway, being the major signalling pathway regulating the expression of the CYP19A1 gene in the ovaries [36][93]. The above-mentioned observations indicate that the benefits of metformin may also be mediated by the inhibition of oestrogen production in the ovaries by the same actions as outlined in ESCs.
In stromal cells of human breast adipose tissue, metformin suppresses the nuclear translocation of CREB-regulated transcription co-activator 2 (CRTC2), which elevates aromatase expression by binding to CYP19A1 PII. It is also the main purpose of AMPK [23][24][80,81]. In granulosa cells, metformin decreases the FSH-induced phosphorylation of CREB, thereby reducing the CREB action, which may cause interference of the CREB-binding protein (CBP)-CRTC2 co-activator complex that connects to CRE in PII of the CYP19A1 gene. Metformin is able to disturb the CREB-CRTC2 complex in human ESCs [20][77]. The above results confirm that metformin attenuates the PGE2-stimulated binding of CREB to CYP19A1 PII, possibly by interfering with the CREB-CRTC2 complex [7][65].
It has been confirmed that metformin interferes with the pathophysiology of endometriosis by reducing matrix MMPs expression, although it is not the major mechanism of its action [37][94]. MMPs are proteolytic enzymes that participate in the reduction and reconstruction of the extracellular matrix. The disturbance of the regulation of MMPs is considered crucial in the progress of pathological disorders such as endometriosis [38][39][40][95,96,97]. Accordingly, MMP inhibitors may impair the progression of the disease.
It has been proven that oxidative status balance may affect lipid and glucose metabolism [41][98]. It has also been confirmed that patients affected by endometriosis have abnormalities in both types of these metabolisms. For example, McKinnon et al. [42][99] reported that in endometrial lesions, the expression of the glucose-4 transporter (GLUT4) protein is increased, possibly due to elevated glucose availability in the growing lesions. Melo et al. [43][100] found that patients with endometriosis often have an unfavorable lipid profile (decrease in high-density lipoprotein and increase in low-density), potentially conducive to oxidative stress (by lipid peroxidation), which tends to increase in this group of women.
The unique therapeutic action of metformin in endometriosis is associated with its impact on inflammation, angiogenesis, invasion, and adhesion, as well as apoptosis. The mechanisms of action of metformin in the above-mentioned processes require a more detailed description.
2.1. The Impact of Metformin on Inflammation
Inflammation plays an important role in the process of endometriosis [44][45][1,101] and is associated with progesterone resistance. Many studies have revealed that metformin suppresses inflammation. In endometrial stromal cells, metformin may limit the inflammatory process by affecting the secretion of IL-6 and IL-8. Metformin inhibits the production of IL-1β and suppresses the production of IL-8. Moreover, human ESCs cultures incubated with metformin show statistically significantly reduced IL-1β-induced IL-8 production in a dose-dependent fashion [46][102]. The same phenomenon was found in the ability of stromal cells to transform androstenedione into estrone (reaction dependent on aromatase activity) as well as in deoxyribonucleic acid (DNA) synthesis (a marker of cell proliferation). The anti-proliferative effect of metformin on IL-8 depends on the cell type because it does not suppress the secretion of IL-8 from eutopic (normal) endometrial stromal cells.
Metformin may reduce serum levels of TNF-α in women with endometriosis [47][103]. Treatment with metformin can intensify the autophagy process by inhibiting mTOR [48][49][104,105]. Co-therapy of quercetin and metformin significantly inhibits mTOR messenger ribonucleic acid (mRNA) expression and increases the gene expression of autophagy factors in ectopic endometrial tissues. This combined treatment regressed endometrial implants in rodents mostly through antiestrogenic and anti-inflammatory functions.
2.2. The Impact of Metformin on Angiogenesis
3.2. The Impact of Metformin on Angiogenesis
Angiogenesis is a physiological phenomenon of blood vessel formation on the basis of the existing ones. This process requires an equilibrium between stimulatory and inhibitory signals. Once the balance is disturbed, the vascularization could be activated. Angiogenesis plays an important role in the neoplastic development and metastases [50][51][106,107]. In vitro and in vivo studies revealed that metformin could inhibit tumour angiogenesis in different mechanisms, such as the AMPK/mTOR pathway activation, downregulation of platelet-derived growth factor B (PDGF-B), and inhibition of several angiogenic-related proteins, such as vascular endothelial growth factor (VEGF), hypoxia-inducible factor-1 (HIF-1), insulin-like growth factor binding protein-2 (IGFBP-2), platelet-derived growth factor-AA (PDGF-AA), VEGF, angiogenin, matrix metalloprotein-9 (MMP-9), and endostatin [52][53][54][55][56][108,109,110,111,112]. In vivo, metformin limited the microvascular tumour density and modified the perivascular/endothelial cell ratio [57][113].
Although endometriosis is a benign condition, it is linked to various cancer-related processes including angiogenesis [58][59][114,115]. It has been reported that angiogenesis resulting in the dysfunction of the vascular basal membranes, as well as the surrounding extracellular matrix (ECM) [60][116], is a hallmark of endometriosis. Unfortunately, precise angiogenesis mechanisms in this disorder are still unknown [58][61][114,117]. The use of metformin prevents the progression of endometrial lesions. Metformin suppresses the proliferation of endothelial cells and reduces vasculature lesions by inhibiting the expression of VEGF. Yilmaz et al. noticed that in rats treated with metformin, the concentrations of superoxide dismutase (SOD) and MMP-2 were increased, whereas the levels of VEGF and MMP-9 were decreased in the endometrial lesions [62][118].
It is worth noticing that metformin decreased the risk of endometrial hyperplasia by reducing the expression levels of urothelial cancer associated 1 (UCA1), transforming growth factor-β (TGF-β) and protein kinase B, while increasing the levels of microRNA-144 and active Caspase-3 [63][119].
2.3. The Impact of Metformin on Adhesion and Invasion
Metformin may inhibit the vascular adhesion molecule expression in endometriosis. Metformin may affect the binding of monocytes to endothelial cells by reducing vascular cell adhesion molecule-1 (VCAM-1) expression on activated endothelial cells. Studies confirmed that metformin in rat abdominal endometriosis induced reduction in weight, mean area, and volume of lesions compared to no reduction in the control group [11][64][13,69]. Histological assessment of the lesions proved that metformin therapy was related to a significant decrease in the number of epithelial cells. The risk and severity of adhesions were also lower.
Lipolysis-stimulated lipoprotein receptor (LSR) revealed itself as a recent molecular constituent of tricellular contacts that have a barrier mechanism for the cellular sheet [65][120]. LSR recruits tricellulin (TRIC), which is the first molecular element of tricellular tight junctions. The knockdown of LSR enhances invasion and motility of some cancer cells [66][121]. In endometriosis, LSR is noticed in the secretory phase of normal endometrial epithelial cells. Furthermore, LSR in cancer is reduced in association with the malignancy. The downregulation of LSR by siRNA provokes cell migration, proliferation and invasion, while TRIC transfers from the tricellular region to the bicellular region of the membrane [67][122]. Metformin increases LSR expression and prevents the migration and invasion of the cells activated by knockdown of LSR in those treated with siRNA or leptin in endometrial cancer cells. Shimada et al. noticed upregulation of LSR by metformin via MAPK and upregulation of LSR via MAPK, PI3K and JAK2/STAT [67][122].
2.4. The Impact of Metformin on Apoptosis
Homeostasis of tissues is modulated by apoptosis. A balance between cell proliferation and apoptosis preserves this homeostasis against cellular disturbances. In endometriosis, the reduction in cell death can lead to the progression of this disease [68][69][123,124]. The ratio of cell apoptosis is suppressed in endometrial cells [70][125]. Moreover, in endometriosis, the induction of the NF-κB pathway is associated with both apoptosis and proliferation [71][72][126,127].
Hsieh Li et al. [73][128] observed that metformin reduces p53 expression levels and significantly induces apoptotic cell death. Contrary to this, Xiao et al. [74][129] noticed that metformin increases AMPK stimulation without modifying the expression levels of p53 and liver kinase B1 (LKB1). Metformin is able to increase LKB1 phosphorylation, promote p53 activation and AMPK, and suppress cell cycle progression [75][130]. Furthermore, Chen et al. [76][131] demonstrated that cytotoxicity induced by metformin occurs through the induction of the caspase-dependent apoptotic signalling pathway.
Metformin has inhibitory actions on cell proliferation, metastasis, apoptosis, angiogenesis, and chemoresistance in diverse malignancies in vivo and in vitro, such as ovarian, hepatocellular, and endometrial cancers [77][78][79][132,133,134]. The above inhibitory properties of metformin are associated with the induction of the PI3K/AKT/mTOR signalling pathway [77][78][79][132,133,134]. Metformin also suppresses tumour growth by stimulating the ATM serine/threonine kinase/AMPK/p53 signalling pathway and reducing the AKT/mTOR/eukaryotic translation initiation factor 4E-binding protein 1 signalling pathway. It leads to an increased response to radiation [80][135]. Moreover, clinical studies confirmed that metformin use is associated with higher overall survival in the group of patients with various cancers [81][82][136,137]. Metformin, by acting on the AMPK/p53 and PI3K/AKT/mTOR signalling pathways, may induce apoptosis and cell cycle arrest.
To summarize, metformin can reduce angiogenesis, inflammation, invasion, and adhesion and may cause regression of endometrial lesions. Insulin sensitizers are reported to diminish cytokine and chemokine expression in endometriotic stromal cells [83][138], regulate angiogenesis [84][53], and provoke apoptosis in endometrial lesions [85][57].