As human life expectancy is rising, the incidence of age-associated diseases will also increase. Scientific evidence has revealed that healthy diets, including good fats, vitamins, minerals, or polyphenolics, could have antioxidant and anti-inflammatory activities, with antiaging effects. Recent studies demonstrated that vitamin K is a vital cofactor in activating several proteins, which act against age-related syndromes. Thus, vitamin K can carboxylate osteocalcin (a protein capable of transporting and fixing calcium in bone), activate matrix Gla protein (an inhibitor of vascular calcification and cardiovascular events) and carboxylate Gas6 protein (involved in brain physiology and a cognitive decline and neurodegenerative disease inhibitor).
1. Introduction
Aging is a multifactorial process that gradually deteriorates the physiological functions of various organs, including the brain, musculoskeletal, cardiovascular, metabolic, and immune system leading to numerous pathological conditions with high rates of morbidity and mortality. Oxidative stress (OS) and chronic inflammation are fundamental pathophysiological mechanisms in the aging progression
[1][2][3][1,2,3].
As human life expectancy is rising, age-related diseases will increase as well. Recent studies validated the importance of modifiable lifestyle factors, diet included, in the attenuation of pathological changes in mature adults
[4]. Healthy fats, vitamins, minerals, polyphenolics, with antioxidant and anti-inflammatory activity, can increase the quality of life and influence the aging process, and among these factors, vitamin K (VK) has an important part
[5].
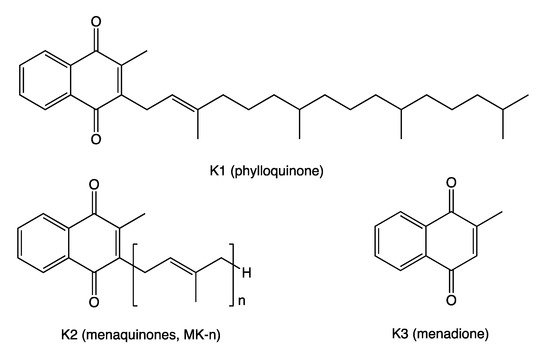
VK is known for its role in synthesizing some blood-clotting proteins (K for koagulation in German). VK represents a fat-soluble family of compounds with a common chemical structure, a 2-methyl-1,4-naphthoquinone ring and a variable aliphatic side-chain. The variable aliphatic chain differentiates two isoforms: vitamin K1 (VK1) or phylloquinone (PK) and vitamin K2 (VK2), usually designated as menaquinone (MK). MK exists in multiple structures, which are distinguished by the number of isoprenyl units and saturation in the side-chain (MK-n, where n is the number of isoprenyl units)
[6]. These acronyms were used interchangeably throughout this
article
ntry. The most common subtypes in humans are the short-chain MK-4, which is the only MK produced by systemic conversion of phylloquinone to menaquinone, and MK-7 through MK-10, which are synthesized by bacteria. VK3 (menadione), without side-chain and classified as a pro-vitamin, is a synthetic form of this vitamin (
Figure 1).
Figure 1. Vitamin K chemical structures. Vitamins K1 (VK1), K2 (VK2), and K3 (VK3) share the naphthoquinone ring; VK1 has a phytyl side-chain; VK2 has a side-chain with a varying number of isoprenyl units; VK3 has no side-chain.
Dark green leafy vegetables are the main sources for dietary PK (e.g., collards, turnip, broccoli, spinach, kale), 70–700 μg/100 g, as well as several fruits (e.g., dried prunes, kiwifruit, avocado, blueberries, blackberries, grapes), 15–70 μg/100 g, and some nuts (pine nuts, cashews, pistachios), 10–75 μg/100 g
[7][8][7,8]. In contrast, the main sources of VK2 are fermented foods, cheeses, eggs, and meats (
Table 1)
[9][10][9,10].
Table 1.
Vitamin K2: food category, sources, and amount.
| Food Category |
Food Source |
VK2 * |
| Fermented foods |
Natto
Sauerkraut |
850–1000 (90% MK-7, 8% MK-8)
5.5 (31% MK-6, 23% MK-9, 17% MK-5 and -8) |
| Hard cheeses |
|
50–80 (15–67% MK-9, 6–22% MK-4, 6–22% MK-8) |
| Soft cheeses |
|
30–60 (20–70% MK-9, 6–20% MK-4, 6–20% MK-8) |
| Eggs |
Yolk |
15–30 (MK-4) |
| Meats |
Pork, beef, chicken |
1.4–10 (MK-4) |
*—μg/100 g food sample; MK-n—menaquinone.
Although dietary PK in vegetables is the major source of the VK intake (80–90%), only 5–10% is absorbed, whereas MKs from dairy products are almost completely absorbed. PK, tightly bound to plant chloroplasts, as well as PK digested with some phytochemicals (e.g., saponins, tannins, fibers, phytates) found in pulses, is less bioavailable to human. Though, PK from collards and broccoli is more bioavailable than PK from spinach
[11][12][11,12].
Both VK1 and VK2 are recognized as cofactors for enzyme γ-glutamyl carboxylase (GGCX), which converts glutamic acid (Glu) to a new amino acid γ-carboxyglutamic acid (Gla), in VK-dependent proteins (VKDPs) during their biosynthesis
[13]. These VKDPs require carboxylation to become biologically active, and the negatively charged γ-carboxyglutamic acid residues have a high affinity for positively charged calcium ions
[14].
VKDPs can be classified as hepatic and extrahepatic. The hepatic VKDPs are largely involved in blood coagulation. Extrahepatic VKDPs perform different tasks: osteocalcin (OC) regulates the bone formation and mineralization, the matrix Gla protein (MGP) is a potent inhibitor of vascular calcification, nephrocalcin is involved in kidney functions, the growth arrest-specific protein 6 (Gas6) in the development and differentiation of nervous system
[15]. Additionally, some extrahepatic VKDPs (proteins C and S) inhibit coagulation by inactivating specific coagulation factors necessary to form blood clots
[16].
Recent findings revealed the novel role of VK as an antioxidant and implicitly anti-inflammatory agent independent of its GGCX cofactor activity
[17]. The antioxidant properties of VK are based on a protective action against oxidative cellular damage and cell death by (1) direct reactive oxygen species (ROS) uptake
[17]; (2) the limiting of free radical intracellular accumulation
[18], and (3) inhibition of the activation of 12-lipoxygenase
[19].
Scientific evidence suggests that VK also has anti-inflammatory activity, a vital component against various chronic aging diseases
[20]. VK inhibits the activation of the nuclear factor kappa B (NF-кB) and thus decreases the production of proinflammatory cytokines
[17]. VK is significantly and inversely related to individual inflammatory biomarkers and inflammatory processes due to its anti-inflammatory effects
[21].
The daily reference intake of VK is based mostly on bleeding-associated studies, and it varies between countries. US dietary guidelines recommend daily intakes of 90 and 120 μg for women and men, respectively, while the guidelines in the United Kingdom are set at 1 μg/kg body weight/day
[11]. However, these recommendations are insufficient to induce complete carboxylation of all VKDPs. Only MK-7, having higher bioavailability and longer half-life, proved to promote γ-carboxylation of extrahepatic VKDPs at the current recommended levels, while the recommended levels of both PK and MK-4 have been shown to decrease γ-carboxylation of VKDPs
[22].
Based on estimated dietary consumption, PK accounts for 50%, MK-4 makes up 10%, and MK-7, -8, and -9 represent 40% of total absorbed VK
[23]. Being a fat-soluble vitamin, VK is taken up in the small intestine in the presence of dietary fat. A key mediator of intestinal VK absorption is Niemann–Pick C1-like 1 (NPC1L1) protein, a cholesterol and phytosterol transporter found in enterocytes and hepatocytes
[24][25][24,25]. After absorption, PK is delivered to the liver and other tissues. It can be used unchanged, or it may be metabolized by certain types of microbiota into VK2 or into menadione in the human intestinal cells. A portion of menadione is transformed to MK-4, the dominant MK form in animal tissues
[26]. However, there are tissue-specific VK distribution patterns. PK was found in all tissues with relatively high levels in the liver and heart but lower levels in the brain, lung and kidney. Compared to PK, MKs seem to be more important for extrahepatic tissues
[27]. MK-4 levels were high in the brain and kidney and low in the liver, heart and lungs. The increased quantities of MK-4 in the brain suggest that this K vitamin is the active form of VK in this region
[28]. Growing evidence advocates that MK-4 has a number of biological functions, including promoting growth factor of neuron-like cells, mediating apoptosis in several cancer cells, controlling glucose homeostasis
[29]. In the central nervous system (CNS), MK-4 controls the activity of proteins involved in tissue renewal and cell growth control, myelination, mitogenesis, chemotaxis, neuroprotection
[30]. The medium and long side-chain MKs were recovered mostly in the liver samples
[31]. MK-7 and MK-4 converted from MK-7 increase collagen production and bone mineral density, promoting bone quality and strength
[17]. As VK1, MK-4, and MK-7 have distinct bioavailability and biological activities, their recommended levels should be established based on their relative activities
[32].
2. Vitamin K in Bone Health
The musculoskeletal system, comprised primarily of muscle and bone, and the adipose tissue are connected through biological mechanisms underlying the physiological and pathophysiological crosstalk among muscle, bone, and fat
[17]. Thus, several myokines (interleukin-6 (IL-6), myostatin) secreted by muscle have been identified as having effects on bone. Osteokines, especially OC, has been shown to have an endocrine impact on muscle, while adipokines (leptin, adiponectin, resistin) could act on either muscle or bone
[33][37]. An in vitro study revealed that both carboxylated OC (cOC) and undercarboxylated OC (ucOC) increased secretion of adiponectin and the anti-inflammatory cytokine IL-10 and also inhibited secretion of tumor necrosis factor-α (TNF-α), but only cOC suppressed inflammatory IL-6 cytokine
[34][38].
Thus, modifiable risk factors, such as healthy diets and physical activity, can positively affect these tissues. The role of calcium and vitamin D (vitD) in preventing osteoporosis is well established. However, more recent evidence suggests that other foods, such as fruit and vegetable, may have an essential role in bone health. Physical activity contributes to bone health by increasing serum total OC (tOC) and adiponectin, reducing leptin, and lowering insulin resistance
[35][39].
Bone strength is determined by bone mineral content (BMC) and its quality and is associated with biological senescence and vitamin (B, D, K) deficiencies. As VK activates tissue-specific VKDPs, such as prothrombin, OC, or MGP, via the γ-carboxylation of Glu to Gla molecules, insufficient VKDPs γ-carboxylation is a sensitive, tissue-specific marker of VK deficiency
[36][40]. Several studies revealed that VK is involved in bone metabolism and inhibits bone resorption in a dose-dependent manner. Binkley et al. showed that more than 250 µg/d VK intake is required for γ-carboxylation of OC
[37][41].
Circulation OC is a marker of bone turnover. Of the total amount of OC that is released into the circulation, 40 to 60% is ucOC. This fraction of OC, being sensitive to VK intake, is a marker for VK status, usually revealing a lower VK availability
[38][42]. Low dietary VK consumption and a high proportion of ucOC are independent risk factors for bone fractures in mature populations
[39][40][41][42][43][43,44,45,46,47].
Table 2Table 2. summarizes the studies that showed an association between VK intake and bone parameters in mature subjects.
Table 2.
The effect of VK intake on bone outcome parameters.
| Author, Year, Country [Ref.] |
Subjects (W:M)
Age (Mean ± SD) |
Design (Length) |
Intervention
Exposure |
Findings |
| Shiraki et al. 2000 Japan [40][44] |
241 PMO
67.2 y |
prospective
2 y |
45 mg/d MK-4
vs. control |
↓ ucOC (p < 0.0001)
↑ cOC (p = 0.0081)
↓ fracture risk (p = 0.0273) |
| Iwamoto et al. 2001 Japan [44][48] |
72 PMO
65.3 y |
prospective
2 y |
45 mg/d MK-4 + Ca
vs. Ca |
↓ vertebral fractures (p < 0.0001)
↑ BMD (forearm) (p < 0.0001) |
| Purwosunu et al. 2006 Indonesia [45][49] |
63 PMO
60.8 y |
RCT
48 w |
45 mg/d MK-4 + Ca
vs. Ca |
↓ ucOC (p ˂ 0.01)
↑ BMD (lumbar) (p < 0.05) |
| Bolton-Smith et al. 2007 UK [41][45] |
244 healthy W
68.2 y |
RCT
2 y |
200 μg/d VK1 + 10 μg/d vitD3 + Ca vs. placebo |
↓ ucOC (p < 0.001)
↑ BMD (ultradistal radius) (p < 0.01) |
| Knapen et al. 2007 Netherlands [46][50] |
Norway [80][94]325 PMW
66.0 y |
147 (66:81)
74.0 ± 10 yRCT
3 y |
45 mg/d MK-4
|
20 movs. placebo |
↑ BMC (p < 0.05) and bone strength (femoral neck) |
| VK levels |
| dp-ucMGP |
↓ VK levels |
↑ dp-ucMGP in symptomatic AS |
Booth et al. 2008
USA [47][51] |
Schlieper et al. 2011
Serbia [81][95] | 452 (267:185)
68.4 y |
RCT
3 y |
500 μg/d PK
vs. control |
↓ ucOC (p ˂ 0.0001) |
| 188 (89:99) |
58 ± 15 y |
Follow-up,
1104 days |
VK levels
dp-ucMGP
dp-cMGP |
↓ dp-cMGP
↑ CV: HR = 2.7 (95% CI: 1.2–6.2, p = 0.015)
↑ All-cause: HR = 2.16 (95% CI:
1.1–4.3, p = 0.027) |
Cheung et al. 2008
Canada [48][52] |
Ueland et al. 2011
Norway [82][96] | 400 PMOa
59.1 y |
RCT
2–4 y |
5 mg/d VK1
vs. placebo |
↓ fracture risk ( |
179 (39:140)
56 yp = 0.04) |
| 2.9 y |
VK levels |
| dp-ucMGP |
↓ VK levels; ↑ dp-ucMGP
↑ heart failure: HR=5.62 (95% CI: 2.05–15.46, p = 0.001) |
Hirao et al. 2008 Japan [49][53] |
44 PMW
68.4 y |
prospective
1 y |
| Westenfeld et al. 2011 Germany [83][97] |
103 (48:55)
˃ 60.5 y |
RCT | 45 mg/d VK2 + 5 mg/d alendronate vs. 5 mg/d alendronate |
6 w |
G1–45 µg/d MK-7
G2–135 µg/d MK-7
G3–360 µg/d MK-7↓ ucOC (p = 0.014)
↓ ucOC:cOC (p = 0.007)
↑ BMD (femoral neck) (p = 0.03) |
| ↓ dp-ucMGP by 77–93% G2 and G3 vs. control |
Tsugawa et al. 2008 Japan [50][54] |
Dalmeijer et al. 2012
Netherlands [84][ | 379 W
63.0 y |
prospective
3 y |
high VK1 vs. low VK1 |
98] |
60 (36:24)
59.5 y↓ vertebral fracture risk (p < 0.001) |
| RCT |
| 12 w |
G1–180 μg/d MK-7
G2–360 μg/d MK-7 |
↓ dp-ucMGP by 31% G1 and 46% G2 vs. placebo |
Binkley et al. 2009 USA [42][46] |
| van den Heuvel et al. 2013 Netherlands [85][99 | 381 PMW
62.5 y |
RCT
1 y |
1 mg/d VK1 or 45 mg/d MK-4 vs. placebo |
] |
577 (322:255)
59.9 ± 2.9 y |
Follow-up 5.6 y |
VK levels
↓ ucOC (p < 0.001) for both VK1 and MK-4 groups |
| dp-ucMGP |
↓ VK levels; ↑ dp-ucMGP |
| ↑ CVD: HR=2.69 (95% CI: 1.09–6.62, | p = 0.032) |
Yamauchi et al. 2010 Japan [51][55] |
221 healthy W
60.8 ± 9.5 y |
cross-sectional |
260±85 μg/d VK |
↓ ucOC (p |
Caluwé et al. 2014
Norway [86][ | < 0.0001) |
| ↑ BMD (lumbar) ( | p = 0.015) |
| 100] |
165 (83:82)
70.8 y |
RCT
8 w |
360, 720 or 1080 μg MK-7 thrice weekly |
↓ dp-ucMGP by 17–33–46% |
Je et al. 2011
Korea [52][56] |
78 PMW
67.8 y |
RCT
6 mo |
Liabeuf et al. 2014
France [87][101] |
198 (40:158)
64 ± 8 y | 45 mg/d MK-4 + vitD + Ca vs. vitD + Ca |
↓ ucOC ( | p = 0.008)
↑ BMD (lumbar) (p = 0.049) |
| Cross-sectional |
VK levels |
| dp-ucMGP |
↓ VK levels; ↑ dp-ucMGP
↑ PAC: OR = 1.88 (95% CI: 1.14–3.11, p = 0.014) |
Kanellakis et al. 2012 Greece [53][57] |
173 PMW
62.0 y |
RCT
12 mo |
| Cheung et al. 2015 USA [88][102] |
3401 (2245:1156)
61.9 y |
Follow-up
13.3 y | 100 μg PK or
MK-7 + vitD + Ca
vs. control |
↓ ucOC (p = 0.001) *
↑ BMD (lumbar) (p < 0.05) * |
| ↑ VK daily intake |
↓ CVD mortality: HR = 0.78 (95% CI: 0.64–0.95, | p | = 0.016) |
Knapen et al. 2013 Netherlands [54][58] |
Knapen et al. 2015
Norway [89][103] | 244 PMW
60.0 y |
RCT
3 y |
180 μg/d MK-7
vs. placebo |
244 PMW↓ ucOC (p < 0.001)
↑ BMD (lumbar spine, femoral neck), bone strength (p < 0.05) |
| Jiang et al. 2014 China [55][59] |
213 PMW
64.4 y |
RCT
1 y |
45 mg/d MK-4 + Ca
vs. Ca |
↓ ucOC (p < 0.001)
↑ BMD (lumbar) (p < 0.001) |
| Rønn et al. 2016 Denmark [43][47] |
148 PMOa
67.5 y |
RCT
1 y |
375 µg/d MK-7
vs. placebo |
↓ ucOC (p < 0.05)
↓ ucOC:cOC (p < 0.05)
↑ bone structure (tibia) (p < 0.05) |
| Bultynck et al. 2020 UK [56][60] |
62 (42:20)
80.0 ± 9.6 y |
Prospective |
↑ serum VK |
↓ hip fracture risk |
Moore et al. 2020
UK [57][61] |
374 PMO
68.7 y |
cross-sectional |
↑ serum VK1 |
↓ fracture risk (p = 0.04) |
Sim et al. 2020
Australia [58][62] |
30 (10:20)
61.8 ± 9.9 y |
RCT
12 w |
136.7 μg/d VK |
↓ ucOC and ucOC:tOC (p ≤ 0.01) |
BMC—bone mineral content; BMD—bone mineral density; cOC—carboxylated osteocalcin; M—men; PMW—postmenopausal women; PMO—postmenopausal osteoporosis; PMOa—postmenopausal osteopenia; RCT—randomized controlled trial; SD—standard deviation; tOC—total osteocalcin; ucOC—undercarboxylated osteocalcin; W—women; ↑—increase; ↓—decrease. * for both VK1 and MK-4 groups.
In a study including 221 healthy women, VK intake was significantly and negatively correlated with ucOC
[51][55]. Correspondingly, higher VK consumption was associated with beneficial effects on fracture risk and bone health. Following an increased dietary green leafy vegetable intake by consuming approximately 200 g/d, 30 healthy individuals substantially reduced serum tOC, ucOC, and ucOC:tOC levels, suggesting increased entry of OC into the bone matrix, improvement of bone quality and lower fracture risk
[58][62].
Moore et al. investigated the association between circulating VK1 with fracture risk in a study, including osteoporosis, in postmenopausal women. The results showed that serum VK1 concentrations were significantly higher in the group with fewer fractures and negatively associated with fracture risk
[57][61]. The results of a 3-year study had the same conclusions: subjects with low plasma VK1 concentration had significantly higher susceptibility for vertebral fracture, independently of BMD, compared to the high VK1 group
[50][54].
Postmenopausal women with osteopenia who received 5 mg of VK1 supplementation daily for 4 years had a significantly lower rate of fractures (
p = 0.04)
[48][52].
Besides leafy vegetables, dried plums (
Prunus domestica L.), a rich source of VK1, demonstrated bone-protective effects. In a study of 84 osteopenic, postmenopausal women, 65–79 years of age, daily consumption of 50 g of dried plums for 6 months revealed less total body, hip, and lumbar bone mineral density (BMD) loss compared with that of the control group (
p < 0.05), which can be explained by the ability of dried plums to suppress bone turnover and inhibit bone resorption
[59][63]. Dried plums are rich in VK, potassium and minerals that are important to bone metabolism
[60][64]. Booth et al. assessed the spine and hip BMD change in healthy elderly subjects, and after three years of follow-up, the daily PK supplementation did not present any additional benefit to BMD. However, the level of ucOC, associated with increased risk of bone fracture in older adults, significantly decreased
[47][51]. Similar to the previous study, Emaus et al. observed that the daily intake of 360 µg MK-7 for one year increased cOC and decreased ucOC serum levels (
p < 0.001)
[61][65]. Feskanich et al. showed that women aged 38–74 years with higher daily VK intake had lower serum concentrations of ucOC and a 30% reduction in the risk of hip fracture compared to women with an intake of less than 109 μg VK per day
[62][66]. Equally, the prevalence of VK deficiency was found to be higher in older patients (mean age 80.0) with hip fractures than those without
[56][60].
In an intervention study, the use of 150 μg VK1 per day, in combination with physiological relevant doses of genistein, an important isoflavone
[63][67], vitD, and polyunsaturated fatty acids (eicosapentaenoic and docosahexaenoic acids), could reduce fracture risk, at least at the hip, and prevent osteoporosis in postmenopausal women
[64][68]. On one hand, VK2 supplementation might enhance the efficacy of vitD in bone and muscle health, improve bone quality, and reduce fracture risk in osteoporotic patients. On the other hand, vitD enhanced the carboxylation of OC, thus promoting the incorporation of calcium into the bone matrix and supporting bone metabolism
[65][69]. Increased vitD intake should be accompanied by VK and magnesium supplementation to prevent long-term health risks, including hypercalcemia, a calcium buildup leading to calcification of the blood vessels and eventually osteoporosis. Hypercalcemia is not a vitD hypervitaminosis but rather a VK deficiency and higher serum concentrations of ucOC that inhibit calcium absorption in the bones
[66][70].
3. Vitamin K in the Prevention and Therapy of Vascular Calcification and Cardiovascular Diseases
Aging and several pathologic states, such as obesity, diabetes, or chronic kidney disease (CKD), cause degenerative changes of the vascular walls, including inflammation and vascular calcification (VC), leading to arterial stiffening and increased cardiovascular (CV) morbidity and mortality
[67][81].
Ample evidence has shown that VK deficiency is related to the pathogenesis of VC
[67][68][69][70][81,82,83,84]. VK has been suggested to inhibit VC and protect against cardiovascular disease (CVD) through the activation of VKDPs, such as MGP. To accomplish its potent calcification inhibitory function, MGP, secreted in the inactive form, needs activation (carboxylation), which takes place in the presence of VK. Upon activation, MGP binds calcium with high affinity, thereby inhibiting the VC process
[68][82].
VC, a hallmark of senescence and a strong predictor of CV events, is another chronic inflammatory state induced via the generation of proinflammatory cytokines and mediated by the NF-кB signaling pathway. A high VK status may exert anti-inflammatory effects and prevent VC through antagonizing NF-кB signaling
[69][83]. Growing evidence shows that VK as well as nuclear factor erythroid 2–related factor 2 (Nrf2) signaling could play a vital role in blocking ROS generation, cellular senescence, DNA damage, and inflammaging
[70][84].
In CKD, a pathological condition characterized by osteoporosis, sarcopenia, and increase CVD events
[71][85], VC is widespread even at early stages. Besides careful attention to calcium and phosphate balance, no particular therapy enabling regression or inhibiting the progression of VC existed
[72][86]. Accumulating evidence describes the VC mechanism as an active process involving calcification promoters and inhibitors. The biologically active MGP, highly dependent on VK status, is viewed as a strong inhibitor of vascular elastic fiber damage and VC
[73][87] and also the only factor that can actually reverse the process
[74][88]. The inactive, uncarboxylated form of this protein reflected the deficiency of VK status and has been linked with VC and CV events. Growing scientific data show that VK-dependent MGP could offset age-related wear and tear on the arteries, VC, and CVD development
[75][89].
To date, a number of experiments and observational studies examined the effects of VK supplementation and dietary intake on vascular calcification and CVD (
Table 3) in mature populations.
Table 3.
The effects of VK supplementation on vascular calcification.
| Author, Year, Country (Ref.) |
Subjects (W:M)
Age (Mean ± SD) |
Design (Length) |
Intervention
Exposure |
Findings |
| Geleijnse et al. 2004 Netherlands [76][90] |
4807 (2971:1836)
67.5 y |
7 y |
Q1 ˂ 21.6 μg/d VK2
Q2 21.6–32.7μg/d VK2
Q3 ˃ 32.7 μg/d VK2 |
↓ CHD mortality: RR = 0.43 (95% CI: 0.24–0.77, p = 0.005) Q3 vs. Q1
↓ AC: OR = 0.48 (95% CI: 0.32–0.71, p ˂ 0.001) Q3 vs. Q1 |
Gast et al. 2009
Netherlands [77][91] |
16,057 W
57.0 ± 6.0 y |
Longitudinal
8.1 y |
211.7μg/d VK1
29.1μg/d VK2 |
↓ CHD risk for 10 μg VK2: HR = 0.91 (95% CI: 0.85–1.00, p = 0.04) |
| Shea et al. 2009 USA [78][92] |
388 (235:153)
68 y |
RCT
3 y |
500 μg/d VK1 vs. control |
↓progression of CAC |
| Schurgers et al. 2010 France [79][93] |
107 (43:64)
67 ± 13 y |
18 mo |
VK levels
dp-ucMGP |
↓ VK levels
↑ dp-ucMGP levels with CKD stage |
| Ueland et al. 2010 |
|
| 59.5 ± 3.3 y |
| RCT |
|
| 3 y |
| 180 µg/d MK-7 vs. placebo |
| ↓ Stiffness Index β: −0.67 ± 2.78 vs. +0.15 ± 2.51, | p | = 0.018 |
| ↓ cfPWV: −0.36 ± 1.48 m/s vs. +0.021 ± 1.22 m/s, p = 0.040 |
| Kurnatowska et al. 2015 Poland [90][104] |
42 (20:22)
58 y |
RCT
270 days |
90 μg/d MK-7 + 10 μg/d vitD vs. control |
↑ CAC
↓dp-ucMGP |
Asemi et al. 2016
Iran [91][105] |
66 (31:35)
65.5 y |
RCT
12 w |
180 µg/d MK-7 + 10 µg/d vitD + 1 g/d Ca vs. placebo |
↓ levels of left CIMT (p = 0.02)
↓ insulin (−0.9 vs. +2.6, p = 0.01)
↓ HOMA-IR (−0.4 vs. +0.7, p = 0.01) |
Fulton et al. 2016
UK [92][106] |
80 (36:44)
77 ± 5 y |
RCT
6 mo |
100 µg MK-7 vs. placebo |
↓dp-ucMGP (p < 0.001) |
| Kurnatowska et al. 2016 Poland [93][107] |
38 (17:21)
58.6 y |
RCT
9 mo |
90 μg/d MK-7 + 10 μg/d vitD vs. control |
↓dp-ucMGP by 10.7% |
Sardana et al. 2016
USA [94][108] |
66 (6:60) T2D
62 ± 2 y |
Cross-sectional |
VK levels
dp-ucMGP |
↓ VK levels; ↑ dp-ucMGP
↑ cfPWV (β = 0.40, p = 0.011) |
Aoun et al. 2017
Lebanon [95][109] |
50 (20:30)
71.5 y |
RCT
4 w |
360 μg/d MK-7 |
↓ dp-ucMGP by 86% |
| Brandenburg et al. 2017 Germany [96][110] |
99 (18:81)
69.1 y |
RCT
1 y |
2 mg/d VK1 vs. placebo |
↓ progression of AVC (10.0% vs. 22.0%) |
Shea et al. 2017
USA [97][111] |
1061 (615:446)
74 ± 5 y |
Follow-up
12.1 y |
VK1 levels
dp-ucMGP |
↑ CVD risk in HBP patients (n = 489): HR = 2.94 (95% CI: 1.4–6.13, p ˂ 0.01) |
Puzantian et al. 2018
USA [98][112] |
137 (8:129)
59.6 y |
|
VK levels
dp-ucMGP |
↓ VK levels; ↑ dp-ucMGP
↑ cfPWV (β = 0.21; p = 0.019) |
Dal Canto et al. 2020
Netherlands [99][113] |
601 (303:298)
70 ± 6 y |
Follow-up
7 and 17 y |
↓ VK levels
↓ vitD levels |
↑ LVMI: β = 5.9 g/m2.7
(95% CI: 1.8–10.0 g/2.7)
↑ All-cause mortality: HR = 1.64 (95% CI: 1.12–2.39, p = 0.011) |
| Roumeliotis et al. 2020 Greece [100][114] |
66 (31:35)
diabetic CKD
68.5 ± 8.6 y |
Follow-up
7 y |
VK levels
dp-ucMGP |
↓ VK levels; ↑ dp-ucMGP
↑ CVD mortality: HR = 2.82 (95% CI: 1.07–7.49, p = 0.037) |
Shea et al. 2020
USA [101][115] |
3891 (2154:1737)
65 ± 11 y |
Follow-up
13 y |
↓ VK1 levels |
↑ CVD risk: HR = 1.12 (95% CI, 0.94–1.33)
↑ All-cause mortality |
| Wessinger et al. 2020 USA [102][116] |
60 (11:49) chronic stroke
61.7 ± 7.2 y |
Cross-sectional |
VK dietary intake |
Among stroke survivors, 82% reported consuming below the Dietary Reference Intake for VK |
AC—aortic calcification; AS—aortic stenosis; AVC—aortic valve calcification; CAC—coronary artery calcification; cfPWV—carotid-femoral pulse wave velocity; CHD—coronary heart disease; CIMT—carotid intima-media thickness; CKD—chronic kidney disease; dp-ucMGP —dephosphorylated—undercarboxylated matrix gla protein; CVD—cardiovascular diseases; HF—heart failure; HR—hazard ratio; LVMI—left ventricular mass index; M—men; MK—menaquinone; OR—odds ratio; PAC—peripheral arterial calcification; PMW—postmenopausal women; PMO—postmenopausal osteoporosis; PMOa—postmenopausal osteopenia; RCT—randomized controlled trial; RR—relative risk; SD—standard deviation; W—women; ↑—increase; ↓—decrease.
Several studies demonstrated that higher dietary consumption of VK2 significantly reduced the incidence of VC and coronary heart disease (CHD)
[76][77][90,91]. In these studies, no association between VK1 intake and CHD was detected while controlling for confounders. After monitoring 2987 participants during a median follow-up time of 11 years, only dietary MKs, but not VK1 intake, were significantly associated with a lower risk of CHD
[103][117]. Scientific evidence specified that VK1 mainly carboxylate VK-dependent factors in the liver, while VK2 is responsible for the carboxylation of VKDPs in the extrahepatic tissues
[104][118]. Nonetheless, it was demonstrated that higher doses of VK1, namely 2 mg/d, can also act in extrahepatic tissues and delay the progression of VC
[96][110]. Furthermore, low plasma VK1 status was linked with higher all-cause mortality risk
[101][115] and with an increased risk for CVD in older patients treated for hypertension
[97][111].
VK intake slowed the progression of preexisting coronary artery calcification (CAC), a well-known independent predictor of CVD risk, in asymptomatic older men and women
[78][92]. Moreover, adequate consumption of VK-rich foods has been suggested as both preventing action and prospective adjuvant therapy against atherosclerosis and stroke
[102][116].
A combination of low VK and vitD status is associated with the increased left ventricular mass index, a parameter for cardiac structure, which has been shown to predict higher mortality, as well as the augmented risk of all-cause mortality in older populations
[99][113]. In diabetic patients with stable CHD, combined supplementation with MK-7, vitD, and Ca was associated with a significant reduction in maximum levels of left carotid intima-media thickness (a parameter positively linked with diabetes, blood pressures, lipid profiles, inflammatory cytokines), C-reactive protein (CRP) and malondialdehyde (MDA) levels, and a significant increase in high-density lipoprotein (HDL)-cholesterol levels
[91][105].
A functional VK deficiency is strongly associated with an increase in uncarboxylated VK-dependent protein levels, the hepatic protein induced by vitamin K absence-II (PIVKA-II) and extrahepatic dephosphorylated-uncarboxylated matrix Gla protein (dp-ucMGP)
[85][99]. Scientific findings reported that VK could modulate dp-ucMGP levels and that plasma dp-ucMGP levels decline after VK intake in a dose-dependent manner
[83][86][97,100]. Circulating plasma dp-ucMGP levels augmented progressively in many diseases and were directly correlated with the severity of VC, cardiac function and long-term mortality
[79][80][81][82][93,94,95,96]. Equally, in a study involving 2318 subjects, elevated dp-ucMGP increased the risk of CV (
p = 0.027) and all-cause (
p ≤ 0.008) mortality
[105][119]. Similarly, in diabetes patients with high CV risk, elevated levels of dp-ucMGP and lower levels of total ucMGP (
t-ucMGP) are independently related to the severity of peripheral artery calcification
[87][101]. Moreover, higher dp-ucMGP values were independently associated with carotid-femoral pulse wave velocity (cfPWV) in diabetes and CKD patients and may lead to large arterial stiffening
[94][98][108,112].