Global rise of infections and deaths caused by drug-resistant bacterial pathogens are among the unmet medical needs. In an age of drying pipeline of novel antibiotics to treat bacterial infections, antimicrobial peptides (AMPs) are proven to be valid therapeutics modalities. Direct in vivo applications of many AMPs could be challenging; however, works are demonstrating encouraging results for some of them. In this review article, we discussed 3-D structures of potent AMPs e.g., polymyxin, thanatin, MSI, protegrin, OMPTA in complex with bacterial targets and their mode of actions. Studies on human peptide LL37 and de novo-designed peptides are also discussed. We have focused on AMPs which are effective against drug-resistant Gram-negative bacteria. Since treatment options for the infections caused by super bugs of Gram-negative bacteria are now extremely limited. We also summarize some of the pertinent challenges in the field of clinical trials of AMPs.
- antibiotics
- multidrug resistant (MDR) bacteria
- MDR Gram negative bacteria
- antimicrobial peptides (AMPs)
1. Introduction
2. Polymyxins
Polymyxins are a family of cyclic lipopeptides isolated as early as 1947 from spore-forming Gram-positive bacteria Bacillus polymyxa [51]. Polymyxins are produced from non-ribosomal peptide synthetase system (NRPS) employing enzymes PmxA, PmxB, and PmxC [52]. The chemical structures of polymyxins, polymyxin B (B1, B2), and polymyxin E (colistin), vary slightly from each other (Figure 1A, Table 1). The cyclic structures of both polymyxin B (PMB) and polymyxin E (PME) contain four cationic diamino-butyric acid (Dab) at positions 4, 5, 8, and 9 and a polar residue Thr 10 [53]. PMB contains an aromatic residue D-Phe6, whereas the 6th position in PME is substituted with residue D-Leu.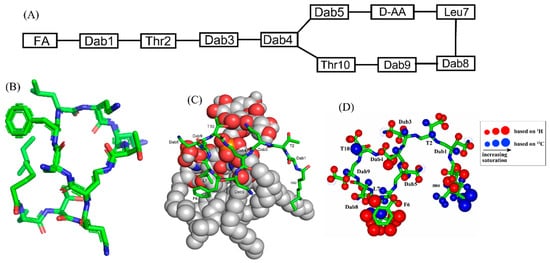
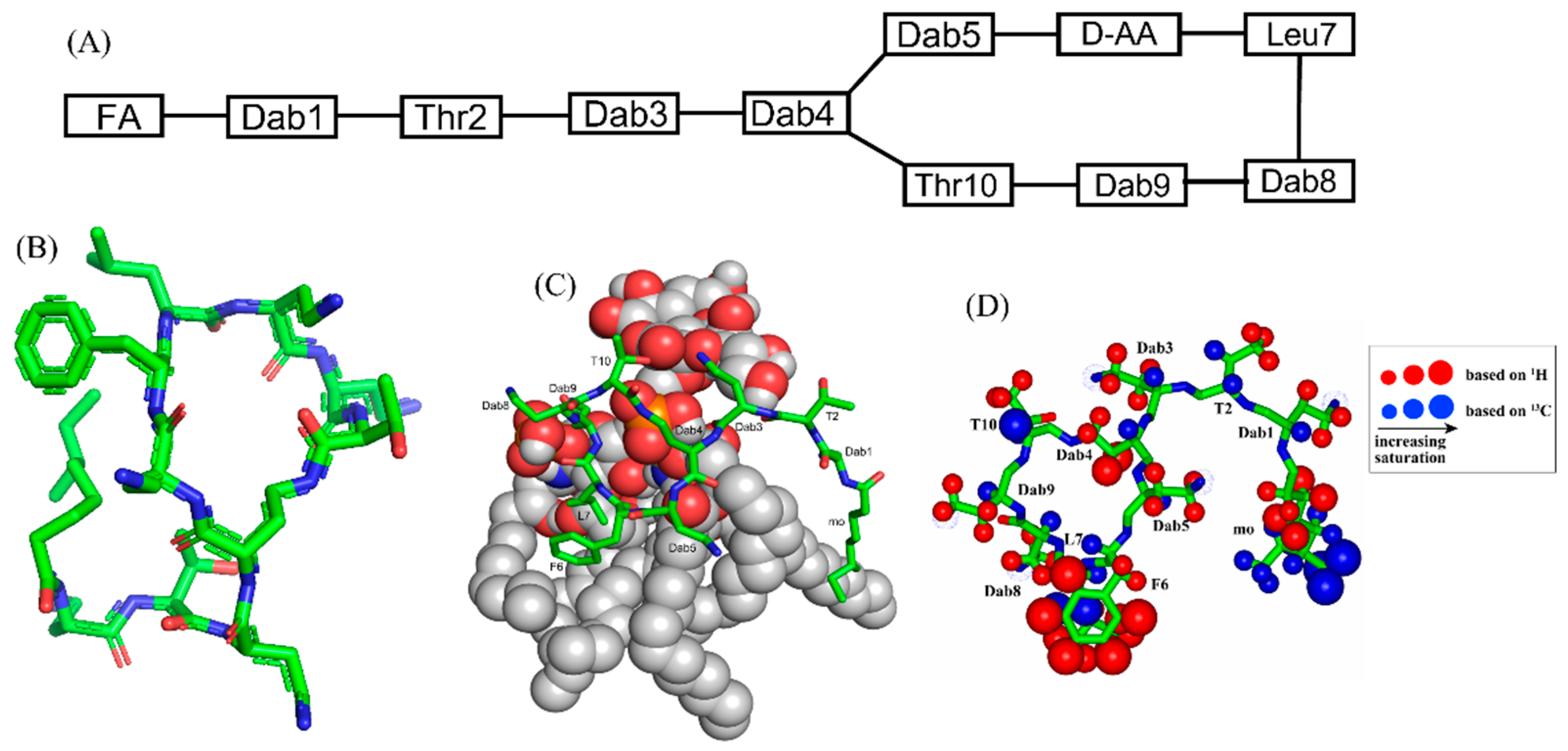 Figure 1. (A) The primary structure of PMB. Both PMB and PME contains cationic Dabs at positions 4, 5, 8, and 9 along with a polar Thr10 residue in their cyclic structures. (B) 3-D structure of PMB represented by a stick model [35]. (C) The docked structure of PMB-LPS complex shows that the cyclic region of PMB (stick model) binds to lipid A moiety of LPS (space filling model) predominantly by ionic/salt bridge interactions. (D) Summary of the epitope mapping of PMB in complex of LPS based on 1H 1D STD experiments (red color) and natural abundance 13C-1H HSQC experiments (blue color) [54]. The structure of PMB is represented as solid stick. The protons are depicted as sphere of different sizes based on determined STD values. (D) is reproduced from reference 54 upon permission obtained from the publisher.
Figure 1. (A) The primary structure of PMB. Both PMB and PME contains cationic Dabs at positions 4, 5, 8, and 9 along with a polar Thr10 residue in their cyclic structures. (B) 3-D structure of PMB represented by a stick model [35]. (C) The docked structure of PMB-LPS complex shows that the cyclic region of PMB (stick model) binds to lipid A moiety of LPS (space filling model) predominantly by ionic/salt bridge interactions. (D) Summary of the epitope mapping of PMB in complex of LPS based on 1H 1D STD experiments (red color) and natural abundance 13C-1H HSQC experiments (blue color) [54]. The structure of PMB is represented as solid stick. The protons are depicted as sphere of different sizes based on determined STD values. (D) is reproduced from reference 54 upon permission obtained from the publisher.
| Compound | FA | Sequence | ||||||
|---|---|---|---|---|---|---|---|---|
| Polymyxin B (PMB) | Methyloctanoyl/methylheptanoyl | Dab1-Thr2-Dab3-cy[Dab4-Dab5-DPhe6-Leu7-Dab8-Dab8-Thr10] | ||||||
| Polymyxin E (PME, Colistin) | Methyloctanoyl/methylheptanoyl | Dab1-Thr2-Dab3-cy[Dab4-Dab5- | DLeu6 | -Leu7-Dab8-Dab8-Thr10] | ||||
| FADD002 | Octanoyl | Dab1-Thr2-Dab3-cy[Dab4-Dab5- | DAda6 | -Leu7-Dab8-Dab8-Thr10] | ||||
| FADD287 | Octanoyl | Dab1-Thr2- | Dap3 | -cy[Dab4-Dab5- | DLeu6 | - | Abu7 | -Dab8-Dab8-Thr10] |
| CA284 | ( | S | )-1-(2-methylpropyl)-piperazine-2-carbonyl+ | Thr1-Dab2-cy[Dab3-Dab4-DPhe-Leu6-Dab7-Dab8-Thr9] | ||||
| SPR206 | (3 | S | )-4-amino-3-(3-chlorophenyl)butanoyl | Thr1-Dab2-cy[Dab3-Dab4-DPhe-Leu6-Dab7-Dab8-Thr9] | ||||
| MicuRx-12 | 3-(2,2-dimethyl-butanoyloxy)-propanoyl (ester bond) | Dab1-Thr2-Dab3-cy[Dab4-Dab5-DPhe6-Leu7-Dab8-Dab8-Thr10] | ||||||
| NAB379 | Octanoyl | Thr1- | DSer2 | -cy[Dab3-Dab4-DPhe5-Leu6-Dab7-Dab8-Thr9] | ||||
| NAB815 | Octanoyl | Dab1-Thr2- | DThr3 | -cy[Dab4-Dab5-DPhe6-Leu7-Abu8-Dab8-Thr10] |
3. β-Sheet AMPs: Protegrins and Thanatin
β-sheet or β-hairpin AMPs are well-folded even in the absence of bacterial membrane, whereas helical AMPs often lack folded conformations in free solution. The stable β-sheet structures are often supported by inter-strand disulfide bond(s). Protegrins (PGs) constitute 16–18-residue long Arg rich AMPs identified from leucocyte of pig (Figure 2). The β-hairpin structures in PGs are maintained by two antiparallel β-strands with disulfide bonds between residues Cys6-Cys15 and Cys8-Cys13. Broad spectrum antibacterial activity under physiological salt solutions had drawn considerable attention for therapeutic developments of PGs [68,69][62][63]. Where PG-1 represents the well-studied member of the group. PG-1 displayed low minimal inhibitory concentration (MIC), ranging between 0.5 to 5 μM, against several strains of Gram-negative and Gram-positive bacteria [68,69,70,71][62][63][64][65]. Iseganan or IB367 is a synthetic variant of PG-1 developed by IntraBiotics Pharmaceuticals through extensive structure activity studies or SAR [72][66]. IB367 was subjected to several phases of clinical trials targeting number of conditions e.g., oral mucositis in cancer patients, prevention of ventilator associated pneumonia, cystic fibrosis and mycoses [73,74][67][68]. However, the status of these clinical trials of iseganan at present is unknown.
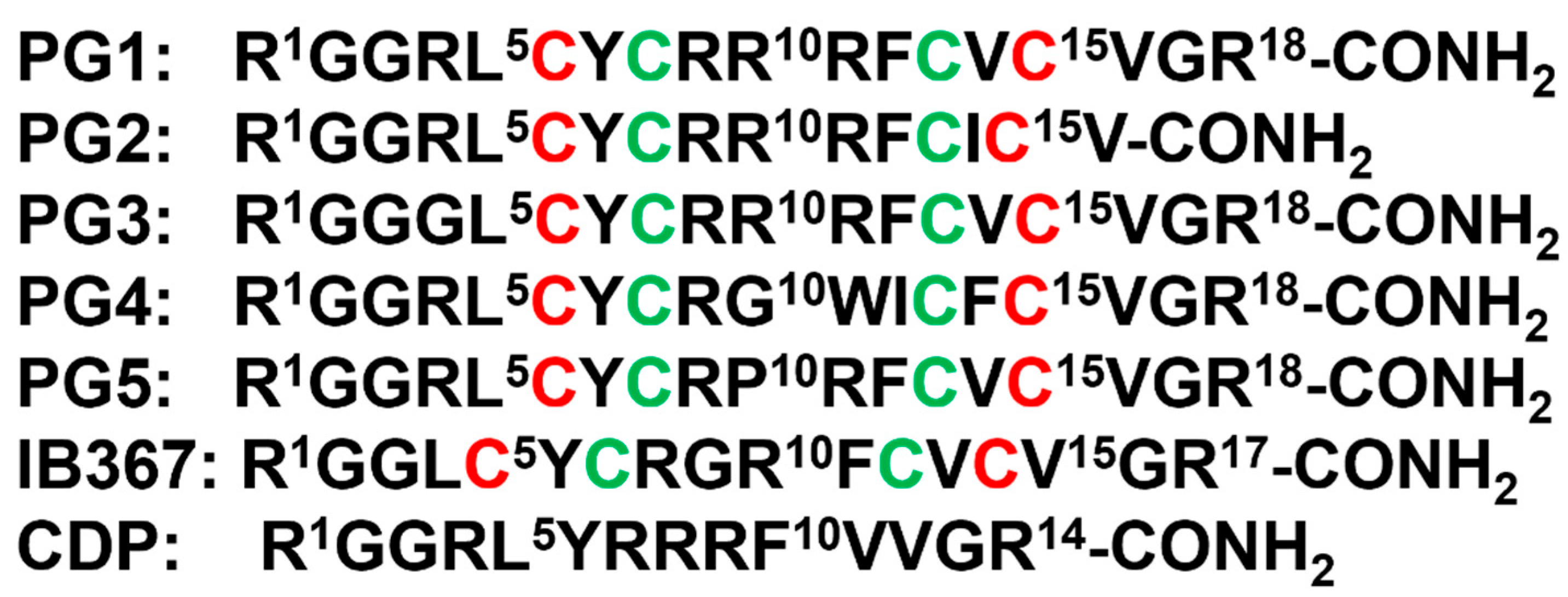 Figure 2. Amino acid sequence of protegrins and its derivatives. The disulfide bonds are formed between Cys6-Cys15 and Cys8-Cys13 residues, depicted by red and green color, respectively.
Figure 2. Amino acid sequence of protegrins and its derivatives. The disulfide bonds are formed between Cys6-Cys15 and Cys8-Cys13 residues, depicted by red and green color, respectively.
4. Outer Membrane Protein Targeting Antibiotics (OMPTA)
OMPTA defines a new class of peptides or peptide mimetics which may be highly effective in killing MDR Gram-negative bacteria [93,94,95][69][70][71]. As a mode of action, OPMTA binds both to LPS and outer membrane proteins resulting in specific Gram-negative activity. In one of these endeavors, protegin-1 was utilized as a starting template generating series of peptidomimetics from designed libraries. In particular, 14-residue long backbone cyclized β-hairpin peptides were developed with inclusion of conserved L-Pro1 and D-Pro14 dipeptide motif [96][72]. Active peptides were screened from these libraries which specifically inhibited only strains of P. aeruginosa with MIC value as low as 0.008 μg/mL [96][72]. One of the candidate peptides termed murepavadin was tested in clinical trials for potential treatment for pneumonia [97][73]. Murepavadin binds to outer-membrane β-barrel protein LptD one of the components of LPS transport machinery [96][72]. However, the exact site of binding of murepavadin or any other related peptides to LptD is not known. As such, LptD has a large, conserved C-terminal domain embedded in LPS outer membrane and a relatively short variable N-terminal at the periplasmic domain [98][74]. The specific P. aeruginosa killing activity of murepavadin over other Gram-negative bacteria may be conferred by binding with the periplasmic domain of LptD. Recently, the phase II clinical trial of murepavadin was halted due to occurrence of acute kidney disease and further preclinical development of the peptide has been undertaken [99][75]. Darobactin, a seven residue (WNWSKSF) peptide, was first isolated from nematode symbiont bacteria Photorhabdus khanii HGB1456. Non-ribosomally synthesized darobactin contains usual sidechain-sidechain covalent crosslinking between indole ring of W1 and β-carbon of W3, β-carbon of K5 and indole ring of W3 [100][76]. Darobactin can kill several Gram-negative bacteria strains in infection animal model but was not effective to tested Gram-positive bacteria. As a mode of action, darobactin directly binds to the outer-membrane protein BamA which is the central component of BamABCDE complex. Binding of darobactin to BamA inhibited chaperon function of the outer membrane protein causing bacterial cell death. BamA/darobactin complex revealed β-sheet like binding of the peptide with the β1-strand of the β-barrel structure of BamA [100,101][76][77].5. MSI Peptides
The first AMP isolated from the anuran family is magainin which was discovered from the skin of a female African clawed frog (Xenopus laevis) by Michael Zasloff in 1987 [102,103][78][79]. Members of the magainin family (magainin-1, magainin-2, and PGLa) are cationic peptides which do not assume any preformed secondary structure in free solution but adopt amphipathic α-helical conformations in membranous environments [104,105][80][81]. Magainins are non-hemolytic and non-cytotoxic host defense peptides that however display potent activity against broad range of bacteria, fungi, and protozoa [14,103][14][79]. Magainin family is made up of two closely similar peptides (magainin-1 and magainin-2), each of which has 23 amino acids and differs by two substitutions at 10th and 22nd position of its primary structure (Figure 53A) [102,103][78][79]. It was also found that magainin-2 has comparatively higher activity than magainin-1. Following this discovery, numerous works were done using magainin-2 and its derivatives to understand activity, structure, and mechanism of action. Through extensive SAR analysis, Zasloff and colleagues at Magainin Pharmaceuticals (now Genaera Corporation) synthesized a 22-residue-long cationic MSI-78 peptide that demonstrated higher potency and greater selectivity to microbial cells compared to human red blood cells [104,106][80][82]. MSI-78 exhibited a broad spectrum of potent antimicrobial activities against both Gram-positive and Gram-negative bacteria including the pathogens associated with the diabetic foot infections (DFI) [106][82]. The MIC50 and MIC90 against all organisms tested from DFI were 16 and 32 μg/mL, respectively [107][83]. Although MSI-78 peptide (known as pexiganan or Locilex) showed promising outcomes against several in vitro, in vivo, and pre-clinical studies, the Food and Drug Administration (FDA) of USA finally denied the new drug application (NDA) for its topical administration as a result of a deficit over a traditional antibacterial medication [108][84]. In 2014, Dipexium Pharmaceuticals initiated another phase III clinical trial using pexiganan and ofloxacin (an FDA approved fluoroquinolone antibiotic) in a comparative clinical study for the same use. However, the trial reported a failure in 2017 since the peptide showed unsatisfactory results at sub-inhibitory concentrations against the tested organisms [109][85]. It is noteworthy to mention that the Locilex (pexiganan topical cream 0.8%) received the advisory from European Medicines Agency (EMA) in 2015 for human and clinical use against DFI.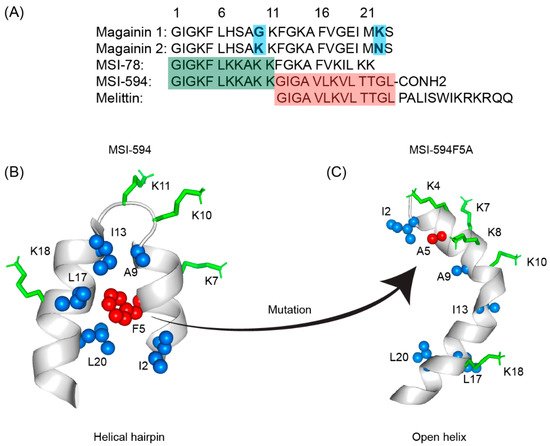
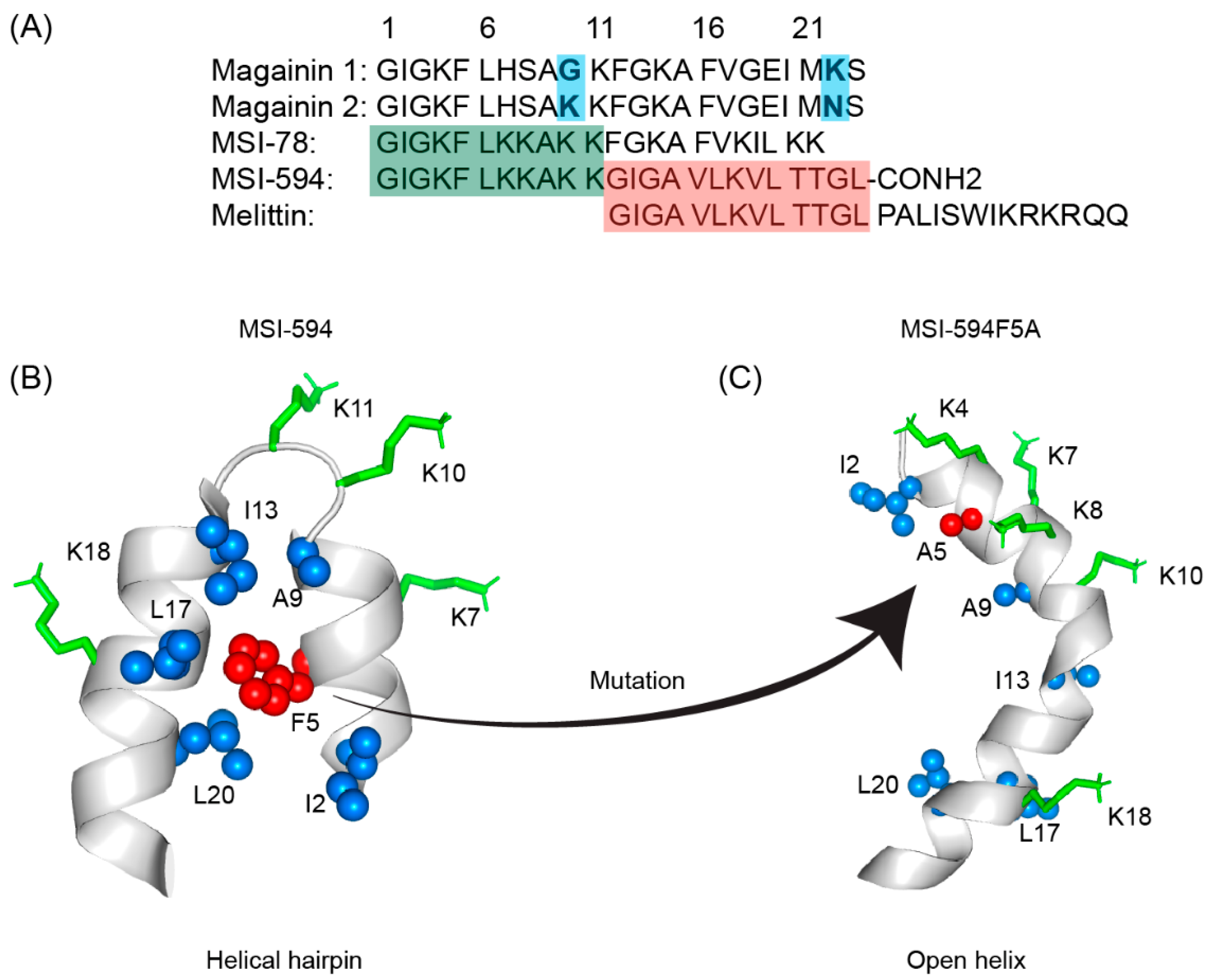 Figure 53. (A) Amino acid sequences of magainin 1, magainin 2, MSI-78, MSI-594, and melittin. The sequence of MSI-594 was derived from MSI-78 and melittin, as indicated by the green and maroon boxes, respectively. (B) MSI-594 adopted helical hairpin or helix-loop-helix structure in presence of LPS, while (C) mutation of Phe5Ala converted the helical hairpin structure to open helix conformation in the same environment [121,122][86][87].
Figure 53. (A) Amino acid sequences of magainin 1, magainin 2, MSI-78, MSI-594, and melittin. The sequence of MSI-594 was derived from MSI-78 and melittin, as indicated by the green and maroon boxes, respectively. (B) MSI-594 adopted helical hairpin or helix-loop-helix structure in presence of LPS, while (C) mutation of Phe5Ala converted the helical hairpin structure to open helix conformation in the same environment [121,122][86][87].
6. LL-37
LL-37 is a cationic α-helical host defense peptide of human that belongs to cathelicidin antimicrobial peptide (CAMP) family [126,127][88][89]. LL-37 is produced by many types of epithelial cells, as well as leukocytes such as monocytes, T cells, B cells, and NK cells and mostly stored in the lysosomes of macrophages and polymorphonuclear leukocytes (PMNs) [127][89]. All cathelicidin AMPs are synthesized as preproproteins (18 KDa) containing a highly conserved N-terminal domain (13.5 KDa) and a vastly diverse antimicrobial domain at the C-terminus (4.5 KDa) [127][89]. The N-terminal domain is typically 94–114 amino acids long and shares sequence homology with cathelin, a cysteine protease inhibitor identified in swine neutrophils. The C-terminus of human cathelicidin comprises 37-residue long LL-37 peptide (Figure 64A) [128][90]. The mature, functional form of the peptide is released after proteolytic cleavage of the signal and cathelin domains [128,129][90][91]. In 1995, three scientific groups discovered the LL-37 peptide based on the study of highly conserved “cathelin” domain [127,130][89][92]. Beside the cell proliferation, immunomodulation, and other signaling roles, the cationic LL-37 peptide (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) shows excellent activity against a broad range of Gram-positive and Gram-negative pathogens [131,132][93][94]. Since the LL-37 peptide possess several promising therapeutic potentials including the induction of angiogenesis, the ProMore Pharma in Poland completed the phase IIb clinical trial of LL-37 for treatment of venous leg ulcers [132,133][94][95].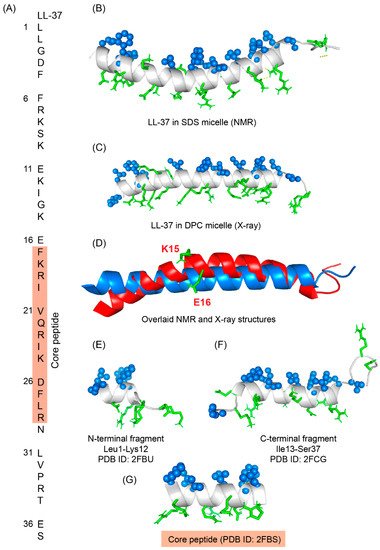
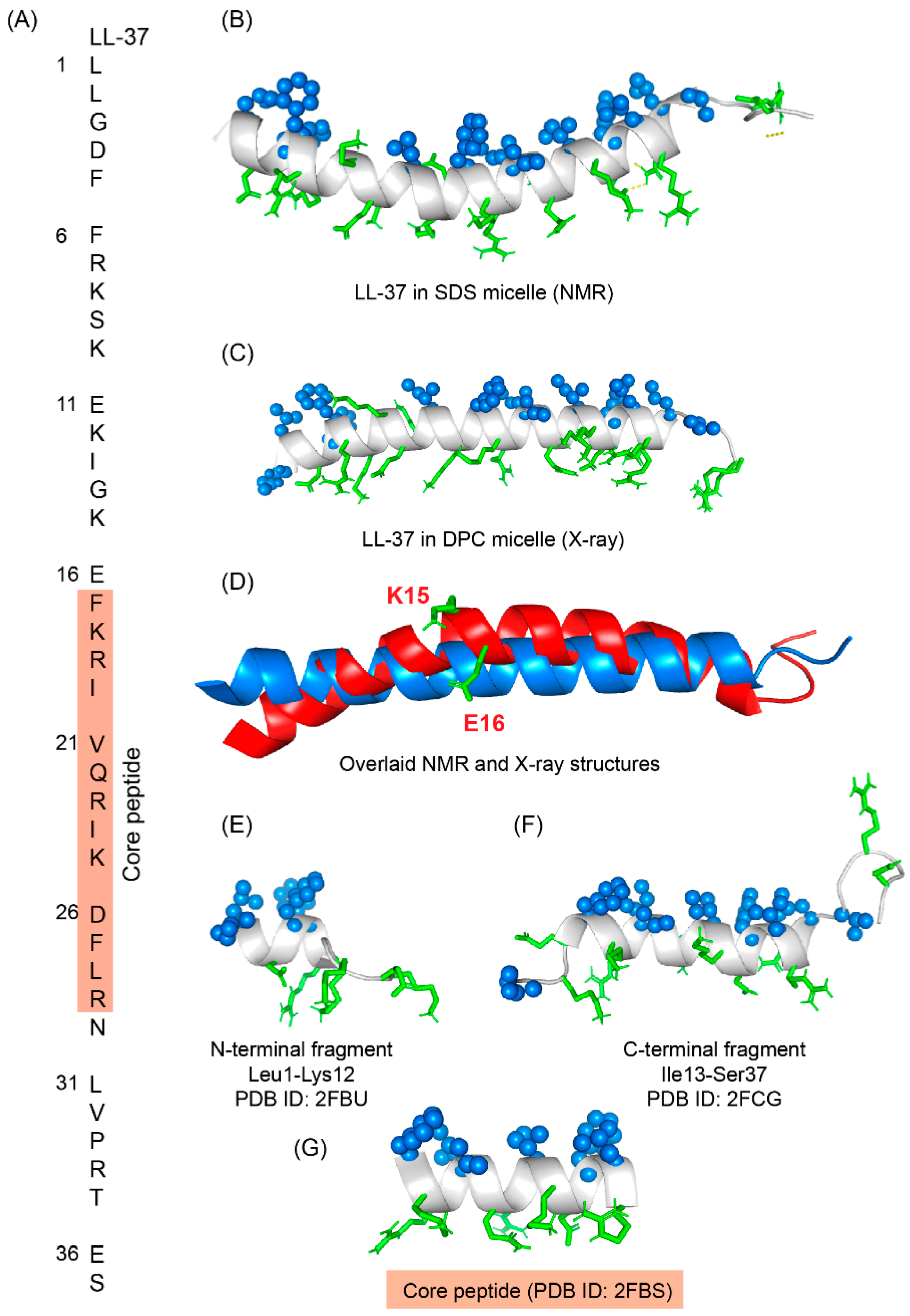 Figure 64. (A) Amino acid sequence of LL-37 peptide; core peptide is highlighted by maroon-colored box. (B) LL37 adopted a twisted amphipathic α-helical conformation in presence of SDS micelle (PDB: 2K6O) [134][96]. (C) In presence of DPC micelle, the peptide obtained a straight helical conformation as seen in the X-ray crystallographic analysis (PDB: 5NMN) [135][97]. (D) The overlaid pdb of NMR and crystallographic structures showed that the SDS bound LL-37 has a kink at residues Lys15 and Glu16. (E–G) The N-terminal, C-terminal, and core peptide fragments also obtained an amphipathic conformation in presence of SDS micelle [136][98].
Figure 64. (A) Amino acid sequence of LL-37 peptide; core peptide is highlighted by maroon-colored box. (B) LL37 adopted a twisted amphipathic α-helical conformation in presence of SDS micelle (PDB: 2K6O) [134][96]. (C) In presence of DPC micelle, the peptide obtained a straight helical conformation as seen in the X-ray crystallographic analysis (PDB: 5NMN) [135][97]. (D) The overlaid pdb of NMR and crystallographic structures showed that the SDS bound LL-37 has a kink at residues Lys15 and Glu16. (E–G) The N-terminal, C-terminal, and core peptide fragments also obtained an amphipathic conformation in presence of SDS micelle [136][98].
References
- Taubes, G. The bacteria fight back. Science 2008, 321, 356–361.
- Kupferschmidt, K. Resistance fighters. Science 2016, 352, 758–761.
- Savage, N. Overcoming resistance. Nature 2020, 586, S55–S56.
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- Antibiotic Resistance Threats in the United States. 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 17 April 2022).
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655.
- Taylor, P.L.; Rossi, L.; De Pascale, G.; Wright, G.D. A forward chemical screen identifies antibiotic adjuvants in Escherichia coli. ACS Chem. Biol. 2012, 7, 1547–1555.
- Theuretzbacher, U. Global antimicrobial resistance in Gram-negative pathogens and clinical need. Curr. Opin. Microbiol. 2017, 39, 106–112.
- Brown, D. Antibiotic resistance breakers: Can repurposed drugs fill the antibiotic discovery void? Nat. Rev. Drug Discov. 2015, 14, 821–832.
- World Health Organization. Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline; World Health Organization: Geneva, Switzerland, 2019.
- Chan, L.W.; Hern, K.E.; Ngambenjawong, C.; Lee, K.; Kwon, E.J.; Hung, D.T.; Bhatia, S.N. Selective Permeabilization of Gram-Negative Bacterial Membranes Using Multivalent Peptide Constructs for Antibiotic Sensitization. ACS Infect. Dis. 2021, 7, 721–732.
- Zgurskaya, H.I.; Löpez, C.A.; Gnanakaran, S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches to Bypass It. ACS Infect. Dis. 2015, 1, 512–522.
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656.
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480.
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230.
- Drayton, M.; Deisinger, J.P.; Ludwig, K.C.; Raheem, N.; Müller, A.; Schneider, T.; Straus, S.K. Host Defense Peptides: Dual Antimicrobial and Immunomodulatory Action. Int. J. Mol. Sci. 2021, 22, 1172.
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215.
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms: My Perspective. Adv. Exp. Med. Biol. 2019, 1117, 3–6.
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9, eaah4680.
- Bhattacharjya, S.; Straus, S.K. Design, Engineering and Discovery of Novel α-Helical and β-Boomerang Antimicrobial Peptides against Drug Resistant Bacteria. Int. J. Mol. Sci. 2020, 21, 5773.
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43.
- Barreto-Santamaría, A.; Arévalo-Pinzón, G.; Patarroyo, M.A.; Patarroyo, M.E. How to Combat Gram-Negative Bacteria Using Antimicrobial Peptides: A Challenge or an Unattainable Goal? Antibiotics 2021, 10, 1499.
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472.
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248.
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self defence? Biochem. Soc. Trans. 2001, 29, 598–601.
- Ebbensgaard, A.; Mordhorst, H.; Aarestrup, F.M.; Hansen, E.B. The Role of Outer Membrane Proteins and Lipopolysaccharides for the Sensitivity of Escherichia coli to Antimicrobial Peptides. Front. Microbiol. 2018, 9, 2153.
- Balhuizen, M.D.; van Dijk, A.; Jansen, J.W.A.; van de Lest, C.H.A.; Veldhuizen, E.J.A.; Haagsman, H.P. Outer Membrane Vesicles Protect Gram-Negative Bacteria against Host Defense Peptides. mSphere 2021, 6, e0052321.
- Bhunia, A.; Domadia, P.N.; Bhattacharjya, S. Structural and thermodynamic analyses of the interaction between melittin and lipopolysaccharide. Biochim. Biophys. Acta BBA Biomembr. 2007, 1768, 3282–3291.
- Krishnakumari, V.; Binny, T.M.; Adicherla, H.; Nagaraj, R. Lipopolysaccharide Modulates Biological Activities of Human-β-Defensin Analogues but Not Non-Ribosomally Synthesized Peptides. ACS Omega 2020, 5, 6366–6375.
- Bhattacharjya, S. NMR Structures and Interactions of Antimicrobial Peptides with Lipopolysaccharide: Connecting Structures to Functions. Curr. Top. Med. Chem. 2016, 16, 4–15.
- Brown, P.; Abdulle, O.; Boakes, S.; Divall, N.; Duperchy, E.; Ganeshwaran, S.; Lester, R.; Moss, S.; Rivers, D.; Simonovic, M.; et al. Influence of Lipophilicity on the Antibacterial Activity of Polymyxin Derivatives and on Their Ability to Act as Potentiators of Rifampicin. ACS Infect. Dis. 2021, 7, 894–905.
- Baker, K.R.; Jana, B.; Hansen, A.M.; Nielsen, H.M.; Franzyk, H.; Guardabassi, L. Repurposing Azithromycin and Rifampicin Against Gram-Negative Pathogens by Combination with Peptidomimetics. Front. Cell. Infect. Microbiol. 2019, 9, 236.
- Bernal, P.; Molina-Santiago, C.; Daddaoua, A.; Llamas, M.A. Antibiotic adjuvants: Identification and clinical use. Microb. Biotechnol. 2013, 6, 445–449.
- Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016, 24, 862–871.
- Swarbrick, J.D.; Karas, J.A.; Li, J.; Velkov, T. Structure of micelle bound cationic peptides by NMR spectroscopy using a lanthanide shift reagent. Chem. Commun. 2020, 56, 2897–2900.
- Ilyas, H.; Kim, J.; Lee, D.; Malmsten, M.; Bhunia, A. Structural insights into the combinatorial effects of antimicrobial peptides reveal a role of aromatic-aromatic interactions in antibacterial synergism. J. Biol. Chem. 2019, 294, 14615–14633.
- Saravanan, R.; Holdbrook, D.A.; Petrlova, J.; Singh, S.; Berglund, N.A.; Choong, Y.K.; Kjellström, S.; Bond, P.J.; Malmsten, M.; Schmidtchen, A. Structural basis for endotoxin neutralisation and anti-inflammatory activity of thrombin-derived C-terminal peptides. Nat. Commun. 2018, 9, 2762.
- Yu, H.Y.; Chen, Y.A.; Yip, B.S.; Wang, S.Y.; Wei, H.J.; Chih, Y.H.; Chen, K.H.; Cheng, J.W. Role of β-naphthylalanine end-tags in the enhancement of antiendotoxin activities: Solution structure of the antimicrobial peptide S1-Nal-Nal in complex with lipopolysaccharide. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1114–1123.
- Kushibiki, T.; Kamiya, M.; Aizawa, T.; Kumaki, Y.; Kikukawa, T.; Mizuguchi, M.; Demura, M.; Kawabata, S.; Kawano, K. Interaction between tachyplesin I, an antimicrobial peptide derived from horseshoe crab, and lipopolysaccharide. Biochim. Biophys. Acta BBA Proteins Proteom. 2014, 1844, 527–534.
- Bai, Y.; Liu, S.; Li, J.; Lakshminarayanan, R.; Sarawathi, P.; Tang, C.; Ho, D.; Verma, C.; Beuerman, R.W.; Pervushin, K. Progressive structuring of a branched antimicrobial peptide on the path to the inner membrane target. J. Biol. Chem. 2012, 287, 26606–26617.
- De Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; de Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; van der Heijde, T.; Boekema, B.K.; et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2018, 10, eaan4044.
- Luther, A.; Urfer, M.; Zahn, M.; Müller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458.
- Nicolas, I.; Bordeau, V.; Bondon, A.; Baudy-Floc’h, M.; Felden, B. Novel antibiotics effective against gram-positive and -negative multi-resistant bacteria with limited resistance. PLoS Biol. 2019, 17, e3000337.
- Mishra, B.; Lakshmaiah Narayana, J.; Lushnikova, T.; Wang, X.; Wang, G. Low cationicity is important for systemic in vivo efficacy of database-derived peptides against drug-resistant Gram-positive pathogens. Proc. Natl. Acad. Sci. USA 2019, 116, 13517–13522.
- Mwangi, J.; Yin, Y.; Wang, G.; Yang, M.; Li, Y.; Zhang, Z.; Lai, R. The antimicrobial peptide ZY4 combats multidrug-resistant. Proc. Natl. Acad. Sci. USA 2019, 116, 26516–26522.
- Spohn, R.; Daruka, L.; Lázár, V.; Martins, A.; Vidovics, F.; Grézal, G.; Méhi, O.; Kintses, B.; Számel, M.; Jangir, P.K.; et al. Integrated evolutionary analysis reveals antimicrobial peptides with limited resistance. Nat. Commun. 2019, 10, 4538.
- Lázár, V.; Martins, A.; Spohn, R.; Daruka, L.; Grézal, G.; Fekete, G.; Számel, M.; Jangir, P.K.; Kintses, B.; Csörgő, B.; et al. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat. Microbiol. 2018, 3, 718–731.
- Silva, A.R.P.; Guimarães, M.S.; Rabelo, J.; Belén, L.H.; Perecin, C.J.; Farías, J.G.; Santos, J.H.P.M.; Rangel-Yagui, C.O. Recent advances in the design of antimicrobial peptide conjugates. J. Mater. Chem. B 2022.
- Kuang, M.; Yu, H.; Qiao, S.; Huang, T.; Zhang, J.; Sun, M.; Shi, X.; Chen, H. A Novel Nano-Antimicrobial Polymer Engineered with Chitosan Nanoparticles and Bioactive Peptides as Promising Food Biopreservative Effective against Foodborne Pathogen. Int. J. Mol. Sci. 2021, 22, 3580.
- Si, Z.; Zheng, W.; Prananty, D.; Li, J.; Koh, C.H.; Kang, E.T.; Pethe, K.; Chan-Park, M.B. Polymers as advanced antibacterial and antibiofilm agents for direct and combination therapies. Chem. Sci. 2022, 13, 345–364.
- Vaara, M. Polymyxins and Their Potential Next Generation as Therapeutic Antibiotics. Front. Microbiol. 2019, 10, 1689.
- Yu, Z.; Qin, W.; Lin, J.; Fang, S.; Qiu, J. Antibacterial mechanisms of polymyxin and bacterial resistance. Biomed. Res. Int. 2015, 2015, 679109.
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure–activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916.
- Bhunia, A.; Bhattacharjya, S. Mapping residue-specific contacts of polymyxin B with lipopolysaccharide by saturation transfer difference NMR: Insights into outer-membrane disruption and endotoxin neutralization. Biopolymers 2011, 96, 273–287.
- Koch-Weser, J.; Sidel, V.W.; Federman, E.B.; Kanarek, P.; Finer, D.C.; Eaton, A.E. Adverse effects of sodium colistimethate. Manifestations and specific reaction rates during 317 courses of therapy. Ann. Intern. Med. 1970, 72, 857–868.
- Gallardo-Godoy, A.; Hansford, K.A.; Muldoon, C.; Becker, B.; Elliott, A.G.; Huang, J.X.; Pelingon, R.; Butler, M.S.; Blaskovich, M.A.T.; Cooper, M.A. Structure-Function Studies of Polymyxin B Lipononapeptides. Molecules 2019, 24, 553.
- Tsuji, B.T.; Pogue, J.M.; Zavascki, A.P.; Paul, M.; Daikos, G.L.; Forrest, A.; Giacobbe, D.R.; Viscoli, C.; Giamarellou, H.; Karaiskos, I.; et al. International Consensus Guidelines for the Optimal Use of the Polymyxins: Endorsed by the American College of Clinical Pharmacy (ACCP), European Society of Clinical Microbiology and Infectious Diseases (ESCMID), Infectious Diseases Society of America (IDSA), International Society for Anti-infective Pharmacology (ISAP), Society of Critical Care Medicine (SCCM), and Society of Infectious Diseases Pharmacists (SIDP). Pharmacotherapy 2019, 39, 10–39.
- Vaara, M.; Vaara, T.; Vingsbo Lundberg, C. Polymyxin derivatives NAB739 and NAB815 are more effective than polymyxin B in murine Escherichia coli pyelonephritis. J. Antimicrob. Chemother. 2018, 73, 452–455.
- Corbett, D.; Wise, A.; Langley, T.; Skinner, K.; Trimby, E.; Birchall, S.; Dorali, A.; Sandiford, S.; Williams, J.; Warn, P.; et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob. Agents Chemother. 2017, 61, e00200-17.
- Bian, X.; Liu, X.; Hu, F.; Feng, M.; Chen, Y.; Bergen, P.J.; Li, J.; Li, X.; Guo, Y.; Zhang, J. Pharmacokinetic/Pharmacodynamic Based Breakpoints of Polymyxin B for Bloodstream Infections Caused by Multidrug-Resistant Gram-Negative Pathogens. Front. Pharmacol. 2021, 12, 785893.
- Vaara, M.; Vaara, T.; Tyrrell, J.M. Structure-activity studies on polymyxin derivatives carrying three positive charges only reveal a new class of compounds with strong antibacterial activity. Peptides 2017, 91, 8–12.
- Gennaro, R.; Zanetti, M. Structural features and biological activities of the cathelicidin-derived antimicrobial peptides. Pept. Sci. 2000, 55, 31–49.
- Dimarcq, J.-L.; Bulet, P.; Hetru, C.; Hoffmann, J. Cysteine-rich antimicrobial peptides in invertebrates. Pept. Sci. 1998, 47, 465–477.
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236.
- Hancock, R.E. Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria. Clin. Infect. Dis. 1998, 27 (Suppl. S1), S93–S99.
- Chen, H.; Gordon, D.; Heinemann, S.H. Modulation of cloned skeletal muscle sodium channels by the scorpion toxins Lqh II, Lqh III, and Lqh alphaIT. Pflüg. Arch. 2000, 439, 423–432.
- Mosca, D.A.; Hurst, M.A.; So, W.; Viajar, B.S.; Fujii, C.A.; Falla, T.J. IB-367, a protegrin peptide with in vitro and in vivo activities against the microflora associated with oral mucositis. Antimicrob. Agents Chemother. 2000, 44, 1803–1808.
- Kollef, M.; Pittet, D.; Sánchez García, M.; Chastre, J.; Fagon, J.Y.; Bonten, M.; Hyzy, R.; Fleming, T.R.; Fuchs, H.; Bellm, L.; et al. A randomized double-blind trial of iseganan in prevention of ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 2006, 173, 91–97.
- Upert, G.; Luther, A.; Obrecht, D.; Ermert, P. Emerging peptide antibiotics with therapeutic potential. Med. Drug Discov. 2021, 9, 100078.
- Luther, A.; Bisang, C.; Obrecht, D. Advances in macrocyclic peptide-based antibiotics. Bioorg. Med. Chem. 2018, 26, 2850–2858.
- Luther, A.; Moehle, K.; Chevalier, E.; Dale, G.; Obrecht, D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 2017, 38, 45–51.
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.; et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013.
- Díez-Aguilar, M.; Hernández-García, M.; Morosini, M.I.; Fluit, A.; Tunney, M.M.; Huertas, N.; Del Campo, R.; Obrecht, D.; Bernardini, F.; Ekkelenkamp, M.; et al. Murepavadin antimicrobial activity against and resistance development in cystic fibrosis Pseudomonas aeruginosa isolates. J. Antimicrob. Chemother. 2021, 76, 984–992.
- Dong, H.; Xiang, Q.; Gu, Y.; Wang, Z.; Paterson, N.G.; Stansfeld, P.J.; He, C.; Zhang, Y.; Wang, W.; Dong, C. Structural basis for outer membrane lipopolysaccharide insertion. Nature 2014, 511, 52–56.
- Dale, G.E.; Halabi, A.; Petersen-Sylla, M.; Wach, A.; Zwingelstein, C. Pharmacokinetics, Tolerability, and Safety of Murepavadin, a Novel Antipseudomonal Antibiotic, in Subjects with Mild, Moderate, or Severe Renal Function Impairment. Antimicrob. Agents Chemother. 2018, 62, e0049018.
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464.
- Kaur, H.; Jakob, R.P.; Marzinek, J.K.; Green, R.; Imai, Y.; Bolla, J.R.; Agustoni, E.; Robinson, C.V.; Bond, P.J.; Lewis, K.; et al. The antibiotic darobactin mimics a β-strand to inhibit outer membrane insertase. Nature 2021, 593, 125–129.
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453.
- Zasloff, M.; Martin, B.; Chen, H.C. Antimicrobial activity of synthetic magainin peptides and several analogues. Proc. Natl. Acad. Sci. USA 1988, 85, 910–913.
- Porcelli, F.; Buck-Koehntop, B.A.; Thennarasu, S.; Ramamoorthy, A.; Veglia, G. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry 2006, 45, 5793–5799.
- Bechinger, B.; Juhl, D.W.; Glattard, E.; Aisenbrey, C. Revealing the Mechanisms of Synergistic Action of Two Magainin Antimicrobial Peptides. Front. Med. Technol. 2020, 2, 615494.
- Islam, K.; Hawser, S.P. MSI-78 Magainin Pharmaceuticals. Idrugs Investig. Drugs J. 1998, 1, 605–609.
- Flamm, R.K.; Rhomberg, P.R.; Simpson, K.M.; Farrell, D.J.; Sader, H.S.; Jones, R.N. In vitro spectrum of pexiganan activity when tested against pathogens from diabetic foot infections and with selected resistance mechanisms. Antimicrob. Agents Chemother. 2015, 59, 1751–1754.
- Gottler, L.M.; Ramamoorthy, A. Structure, membrane orientation, mechanism, and function of pexiganan—A highly potent antimicrobial peptide designed from magainin. Biochim. Biophys. Acta BBA Biomembr. 2009, 1788, 1680–1686.
- Gomes, D.; Santos, R.; Soares, R.S.; Reis, S.; Carvalho, S.; Rego, P.; Peleteiro, M.C.; Tavares, L.; Oliveira, M. Pexiganan in Combination with Nisin to Control Polymicrobial Diabetic Foot Infections. Antibiotics 2020, 9, 128.
- Domadia, P.N.; Bhunia, A.; Ramamoorthy, A.; Bhattacharjya, S. Structure, interactions, and antibacterial activities of MSI-594 derived mutant peptide MSI-594F5A in lipopolysaccharide micelles: Role of the helical hairpin conformation in outer-membrane permeabilization. J. Am. Chem. Soc. 2010, 132, 18417–18428.
- Bhunia, A.; Ramamoorthy, A.; Bhattacharjya, S. Helical hairpin structure of a potent antimicrobial peptide MSI-594 in lipopolysaccharide micelles by NMR spectroscopy. Chemistry 2009, 15, 2036–2040.
- Nijnik, A.; Hancock, R.E. The roles of cathelicidin LL-37 in immune defences and novel clinical applications. Curr. Opin. Hematol. 2009, 16, 41–47.
- Wang, G.; Narayana, J.L.; Mishra, B.; Zhang, Y.; Wang, F.; Wang, C.; Zarena, D.; Lushnikova, T.; Wang, X. Design of Antimicrobial Peptides: Progress Made with Human Cathelicidin LL-37. Adv. Exp. Med. Biol. 2019, 1117, 215–240.
- Thennarasu, S.; Tan, A.; Penumatchu, R.; Shelburne, C.E.; Heyl, D.L.; Ramamoorthy, A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37. Biophys. J. 2010, 98, 248–257.
- Sharma, P.; Sharma, N.; Mishra, P.; Joseph, J.; Mishra, D.K.; Garg, P.; Roy, S. Differential Expression of Antimicrobial Peptides in Streptococcus pneumoniae Keratitis and STAT3-Dependent Expression of LL-37 by Streptococcus pneumoniae in Human Corneal Epithelial Cells. Pathogens 2019, 8, 31.
- Zanetti, M.; Gennaro, R.; Romeo, D. Cathelicidins: A novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995, 374, 1–5.
- Malmsten, M.; Davoudi, M.; Walse, B.; Rydengård, V.; Pasupuleti, M.; Mörgelin, M.; Schmidtchen, A. Antimicrobial peptides derived from growth factors. Growth Factors 2007, 25, 60–70.
- Dürr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta BBA Biomembr. 2006, 1758, 1408–1425.
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111, e24122.
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643.
- Sancho-Vaello, E.; François, P.; Bonetti, E.J.; Lilie, H.; Finger, S.; Gil-Ortiz, F.; Gil-Carton, D.; Zeth, K. Structural remodeling and oligomerization of human cathelicidin on membranes suggest fibril-like structures as active species. Sci. Rep. 2017, 7, 15371.
- Li, X.; Li, Y.; Han, H.; Miller, D.W.; Wang, G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 2006, 128, 5776–5785.