Cancer is a major health problem. Most of the treatments exhibit systemic toxicity, as they are not targeted or specific to cancerous cells and tumors. Specific and targeted therapy can be designed using specific functional modifiers/inhibitors like antibodies, peptides, nanobodies and soluble ligands etc. Another novel therapeutic strategy is by using Oncolytic viruses. These are viruses, which can specifically infect or enter into cancer cells and kill them. Since viruses have evolved natural affinity towards some receptors, their affinity needs to be re-targeted towards cancer cells and de-targeted from their natural receptors. Adenoviruses are very promising gene delivery vectors and have shown immense potential in delivering targeted therapy. Here, we review a wide range of strategies that have been tried, tested, and demonstrated to enhance the specificity of oncolytic viruses towards specific cancer cells. A combination of these strategies and other conventional therapies may be more effective than any of those strategies alone.
- adenovirus vector
- oncolytic adenovirus
- library
- targeting
- promoter
- screening
- High -diversity library
1. Introduction
- Introduction
Cancer is a major health problem, posing a significant burden to individual patients and to society [1][2] [1,2] (https://gco.iarc.fr/). Large efforts have been made to understand its causes and mechanisms of disease progression. Although several advanced therapeutic options based on them are now available, only a few cancer types can be treated effectively if curative surgical resection is not possible [3]. In the vast majority of cases, improving the quality of life of patients even slightly is a practical and significant goal to achieve. Among the available treatments, most of them unfortunately lack cancer specificity, leading to a range of systemic adverse effects that diminish a patient’s quality of life, which is still a big issue [4][3,4]. To improve patient outcomes, researchers have been focused on the development of more cancer-specific, targeted therapies [5][6][7][8][3,5–8].
In general, current strategies of drug development aim to modify the function of a target protein in order to slow down tumor growth or possibly decrease tumor volume. This strategy requires targets to be differentially expressed in tumors, and also functionally important for tumorigenesis and progression [9][10][11][6,9–11]. Numerous high-throughput genomic and proteomic studies comparing healthy and cancerous cells have identified many such potential drug targets [12][13][9,12,13]. These putative targets are then subjected to high-throughput screening with libraries of potential drug candidates, such as peptides, antibodies, natural compounds, chemicals, and aptamers [14][15][16][17][18][19][20][21][22][10,14–22]. Selected molecules that specifically bind to the target are considered for further functional validation [17]. Unfortunately, many potentially druggable genes were found to be difficult to target by this method. Most of these screening experiments showed that despite specific binding of small molecules to tumor targets, the inhibitory or modifying effects of a large fraction of molecules were insufficient to alter their functions and may also exhibit significant toxicity [23][24][17,23,24]. Without strong inhibitory or modifying effects, these molecules cannot be developed for therapy under conventionally with methods [24][25][26][24–26]. Such issues have led to a lack of successful drug candidates [3,9,10,17].
In such situations, tumor targeting by viruses provides an excellent alternative. The natural ability of viruses to interact with cell surface proteins to gain entry into cells makes them attractive tools for targeted therapy [27][28][27,28]. If a virus can be engineered to interact with specific proteins or receptors in a cancerous cell, it can enter the cell to deliver therapeutic cargo or kill the cell by infection inducing cytolysis [29][30][27,29,30]. A major advantage of viruses over small molecules is that the target protein need not be functionally important to the tumor biology. Instead, it must only be specifically expressed or significantly overexpressed in a target cell [29][30][29,30]. Therefore, any gene unique to tumors, irrespective of its functional importance, can be subject to targeting. This dissociation of gene expression from functional relevance eliminates a major limitation, bringing hundreds of genes previously deemed undruggable back into the pool of potential therapeutic targets. This significantly improves the chances of identifying and developing new targeted therapies.
Many viruses cause lysis of infected cells at the end of their infection cycle. Among them, the viruses which are designed to kill cancerous cells are called oncolytic viruses (OVs) [31][32][33][34][27,28,31–34]. Many different viruses have been exploited for this purpose, most notably adenoviruses (AdV) [35], vesicular stomatitis virus (VSV) [36], herpes simplex virus (HSV) [37], vaccinia virus [38], reovirus [39][40][39,40], and Seneca valley virus [41][42][41,42]. Depending on the type of cancer, method of targeting, and therapeutic cargo to be delivered, some viruses may be more suitable than others. Here, we will focus on using adenoviruses as oncolytic viruses and discuss various strategies that have been employed and demonstrated to be effective in achieving a more specific targeting of cancer cells.
2. Adenoviruses as Vectors for Gene Therapy and Oncolytic Viruses
- Adenoviruses as Vectors for Gene Therapy and Oncolytic Viruses
Adenoviruses are popular gene delivery vectors [43]. They can effectively infect both dividing and non-dividing cells [44]. Their double-stranded DNA genome remains episomal, rarely integrating into the host genome [45]. Additionally, while adenoviruses are very common pathogens to humans, they usually cause only mild symptoms in the upper airway, liver, urinary tract, tonsils, enteric, renal, and ocular tissues [12].
Adenoviruses are a family of icosahedral, non-enveloped viruses. Based on serology and genomic sequences, AdVs have been grouped into seven species, each including several types/subtypes [46]. Their capsid is comprised of four structural proteins (hexon, penton, fiber, and pIX), each of which contributes to interaction with the host cell surface. Depending on the type of virus, they can bind to various cell surface proteins to facilitate entry into target cells [47][48][49][47–49]. The knob region of fiber can bind to Coxsackievirus and adenovirus receptor (CAR), Vascular cell adhesion molecule-1 (VCAM), CD80, CD86, MHC1, and Scavenger receptors (SRs) [47]. The shaft of fiber can bind to heparan sulphate proteoglycans (HSPGs) [48] and RGD (Arginine(R)-Glycine(G)-Aspartate(D)) motif in the penton can bind to integrins, CD46, and sialic acid (SA) [47]. In some cases, adenoviral protein interactions can be indirect, mediated by a bridging molecule. Hexon can bind to coagulation factor X (FX) [49], which in turn helps in binding to HSPG. Different serotypes show wide ranges of affinities for these binding partners. There is some discrepancy in determining the most important receptor in a particular cell type, however in most cases several receptors play significant roles. Understanding interactions and affinities between viral proteins and host receptors has helped in designing improved strategies of virus detargeting (abolishing virus affinity towards natural targets) and retargeting (generating affinity towards newer proteins or domains).
AdVs are popular as gene therapy vectors and are the subject of over 100 clinical trials [46]. Among them, Ad2 and Ad5 are the most widely used and widely studied for gene therapy. However, there are some limitations to their use. (1) A significant number of people possess antibodies against common adenoviruses, making it difficult for conventional AdVs to be used for systemic injection. However, in recent years, this thought process has changed and immune response has been somewhat exploited to favor AdV-mediated therapy [50]. (2) AdVs are quickly sequestered to organs such as the liver and lungs after systemic injection. Residential macrophages (e.g., Kupffer cells) play an important role in this process. In one extreme case, high-dose intravascular injection induced a fatal cytokine storm [51]. Erythrocytes and platelets also contribute to sequestration of viral particles upon intravenous delivery [52][53][54][52–54]. In spite of these limitations, an increasing understanding of adenovirus biology has led to designing novel strategies to overcome such limitations, and significant progress has been made.
Detargeting AdVs away from their natural interactions and retargeting of AdVs towards a specific target in an intended cell are very crucial processes in developing a targeted therapy for cancer. Here, we will discuss various strategies employed to achieve detargeting, retargeting, and delivery of novel therapeutic molecules with a few selected examples.
3. Strategies for Specific Targeting of AdV
- Strategies for Specific Targeting of AdV
Broadly speaking, detargeting and specific retargeting can be achieved in multiple ways: (2.1) by selective binding or retargeting towards proteins uniquely expressed or overexpressed on the surface of specific cells; (2.2) by selective expression of effectors (inhibitors or enhancers) in specific cells; (2.3) by inducing conditional or selective replication of viruses in specific cells; (2.4) by combining these methods.
3.1. Selective Retargeting Towards Proteins Uniquely Expressed or Overexpressed on the Surface of Specific Cells
Based on structural and functional studies, we have a decent understanding of how viral proteins interact with host receptors. By altering the sequences of key interacting amino acid residues and domains, their affinity towards natural receptors can be abolished (Section 2.1.1, detargeting) and redirected towards a new protein (Section 2.1.3, retargeting). Since it is difficult to predict how sequence changes may alter the affinity towards an intended target, retargeting towards a specific target is a very challenging process. High-throughput screening to look for such sequences may be the best way (Section 2.1.2, infectivity selective screening for specific targeting) to find retargeting sequences and molecules [55]. Most structural proteins, including hexon, pIX, fiber, and penton, have been exploited by these approaches.
3.1.1 Detargeting of AdV Structural Proteins
Detargeting of Hexon and pIX
Hexon is the most abundant (240 trimers) structural protein in the AdV capsid and is also the main cause of AdV sequestration to the liver [56][57][56,57]. Hexon binds to factor-X (FX), a soluble coagulation factor found in blood plasma, facilitating viral entry into Kupffer cells via scavenger receptors (SR) [58][59][58,59]. Abolishing this interaction will greatly reduce sequestration, allowing circulating AdV to be distributed to other tissues. This can be accomplished by several mechanisms, including by introducing mutations into the hyper variable region (HVR5) of hexon [60], inserting heterologous sequences, swapping the whole HVR [59][61][57,59,61], and by using drugs (such as warfarin and snake venom protein, factor X-binding protein (X-Bp) [62][63][62,63].
Some naturally occurring amino acid sequences (peptides) with known binding characteristics have been used for detargeting and retargeting. A well-known example is the RGD motif found in penton in AdVs[64][65] [64,65]. This peptide is known to interact with integrins that have been slightly modified into RGD-4C and incorporated into HVRs of hexon and the HI-loop of fiber [66][67][68][69][64–69]. In combination with other modifications, this could efficiently detarget fiber from CAR and increase its affinity towards integrin enriched cells. Some studies have shown promising results and progression through clinical trials [70][71][72][73][74][75][70–75]. However, we think looking for naturally occurring motifs and incorporating them into AdV structural proteins to see if they retain their affinity and specificity offers limited options for newly discovered targets of interest.
Another strategy utilizes information from peptide-based screenings that have been performed over the past few decades on key cancer-related drug targets. Such screenings are mostly done by using phage display and/or chemically synthesized libraries. Several potential candidates that specifically bind to individual drug targets have been identified. These peptides, irrespective of their function, can be incorporated into hexon to retarget oncolytic adenoviruses (OAdV). A few successful examples have been described [76][77][64,65,76–78]. Ghosh et al. introduced two such peptides into the HVR5 region of hexon to detarget it away from FX and retarget it towards skeletal muscle cells, which otherwise are not susceptible to AdV binding [78][79][78,79]. It seems that small peptides may be a better choice than larger protein domains, as they may not affect the structural integrity and assembly of the virus [80].
In comparison, altering sequences of hexons seems to be a much safer option than pIX, keeping the structural integrity of capsids in mind. Since hexons and pIX exist in high copy numbers within the virus capsid, a slight increase in their affinity towards an intended target may lead to a stronger cumulative effect on binding [81][82][83][81–83].
Detargeting of Fiber
Fiber is another major AdV structural protein that interacts with host receptors. Fiber consists of two components: (1) the shaft, projecting outward from each apex of the capsid structure; and (2) the knob, located at the distal end of the shaft. The knob structure is stabilized by multiple loops. Structural and mutagenesis studies have shown that the AB-loop of fiber is critical for interaction with CAR, while the c-terminal domain and HI-loop were not involved [84]. Therefore, the HI-loop and C-terminal domain can be manipulated without affecting the fiber’s ability to interact with CAR. Since they are positioned towards the terminus, additional amino acids introduced in these domains may remain out of the core structure of knob, and therefore may not affect its structural stability.
As with hexons, peptides selected by high-throughput screening methods, as mentioned before, have been introduced into the HI-loop or C-terminus to detarget and retarget fiber in AdVs [85][86][66,67,85,86]. A large number of successfully retargeted AdVs towards neuronal cells, brain endothelial cells, vascular endothelial cells, prostate cancer, lung cancer, pancreatic cancer, muscle cells, and receptors-like epidermal growth factor receptor (EGFR) have been reported [85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][66,67,85–99]. Similar to hexon-based studies, keeping the structural stability in mind, small peptides are widely used. However, proteins as large as 83 amino acids have been introduced into this domain, but the viral packaging was somewhat affected [98][99][87,88,98,99]. Therefore, smaller proteins and peptides seem to be the better options.
Simply modifying the HI-loop or c-terminus while keeping the AB-loop intact may not fully abolish fiber–CAR interactions, which is another major reason behind sequestration of AdVs. Hence, complete abolition of fiber–CAR interaction by modifying the AB-loop would be more beneficial. Miura et al. showed improvements in many screening experiments after modifying the AB-loop region [55]. Sato et al. used AB-loop-modified AdV libraries to screen for candidates that specifically bind to CD133. They demonstrated that a peptide sequence “TYMLSRN” introduced into the AB-loop efficiently retargeted the AdV towards CD133 and away from CAR [100]. Similarly, the “VTINRSA” peptide could retarget AdVs to mesothelin [55]. RGD motif and its derivatives were used to modify the fiber, and many such modified viruses are undergoing clinical trials [64–75].
As mentioned above, identifying an appropriate retargeting sequence to replace wild-type sequences is very challenging. A novel alternative to replacing native sequences with retargeting sequences was demonstrated by introducing a “universal acceptor domain” into structural proteins. A bridging molecule can then be used to facilitate the interaction between the virus and its target. The Fc region binding domain of Staphylococcus aureus protein A, biotin acceptor protein (BAP), and a FLAG® peptide have been used as universal acceptor domains [101][102][103][104][105][106][107][108][101–108]. These “universal acceptor domains” are usually large proteins, and their incorporation into structural proteins may affect viral stability. Since the viral fiber protrudes outside the core capsid, we think it is a more suitable location for insertion of large protein domains than hexons which form the core of the capsid.
Swapping of fibers is also an option to change the affinity and evade immune response, at least temporarily [109][110][111][112][113][114][109–114]. The swapping of fibers and chimeric fibers has been very promising in early experiments and clinical trials, especially Ad5/3, Ad5/11, Ad5/9, and Ad5/35 [115][116][117][118][119][95,115–119], Since these fibers (Ad3 and Ad35 fibers) do not bind to CAR, detargeting from CAR becomes an inherent property. However, this may drive their affinity towards desmoglein, CD46, and other native receptors, leading to a different type of off-target effects. This can be overcome by combining this (swapping) with other strategies, such as adding a targeting peptide, to achieve effective detargeting and retargeting [64,68,112].
3.1.2. Infectivity Selective Screening for Specific Targeting: Strategies and Importance
A typical novel drug discovery approach against a specific target mainly relies on screening of a large collection of small molecules and potential drug candidates. Several peptides, antibodies, single-chain fragment variables (ScFVs), nanobodies, and aptamers have been identified by high-throughput screening studies for their ability to bind specifically to a target [120]. Usually, such screenings are done in bacterial systems or in chemical mediums. One of the major concerns is that, the candidates selected under such alien environmental conditions may not retain their properties in eukaryotic cells or animal bodies, where they are ultimately intended to function. Further, they may not remain equally functional and effective when introduced into AdV structural proteins. In most cases, incorporation of such pre-identified motifs into AdV structural proteins affects the viral assembly due to structural deformation. Hence, vast majority of these candidates could not be used for retargeting of AdV [121][79,121].
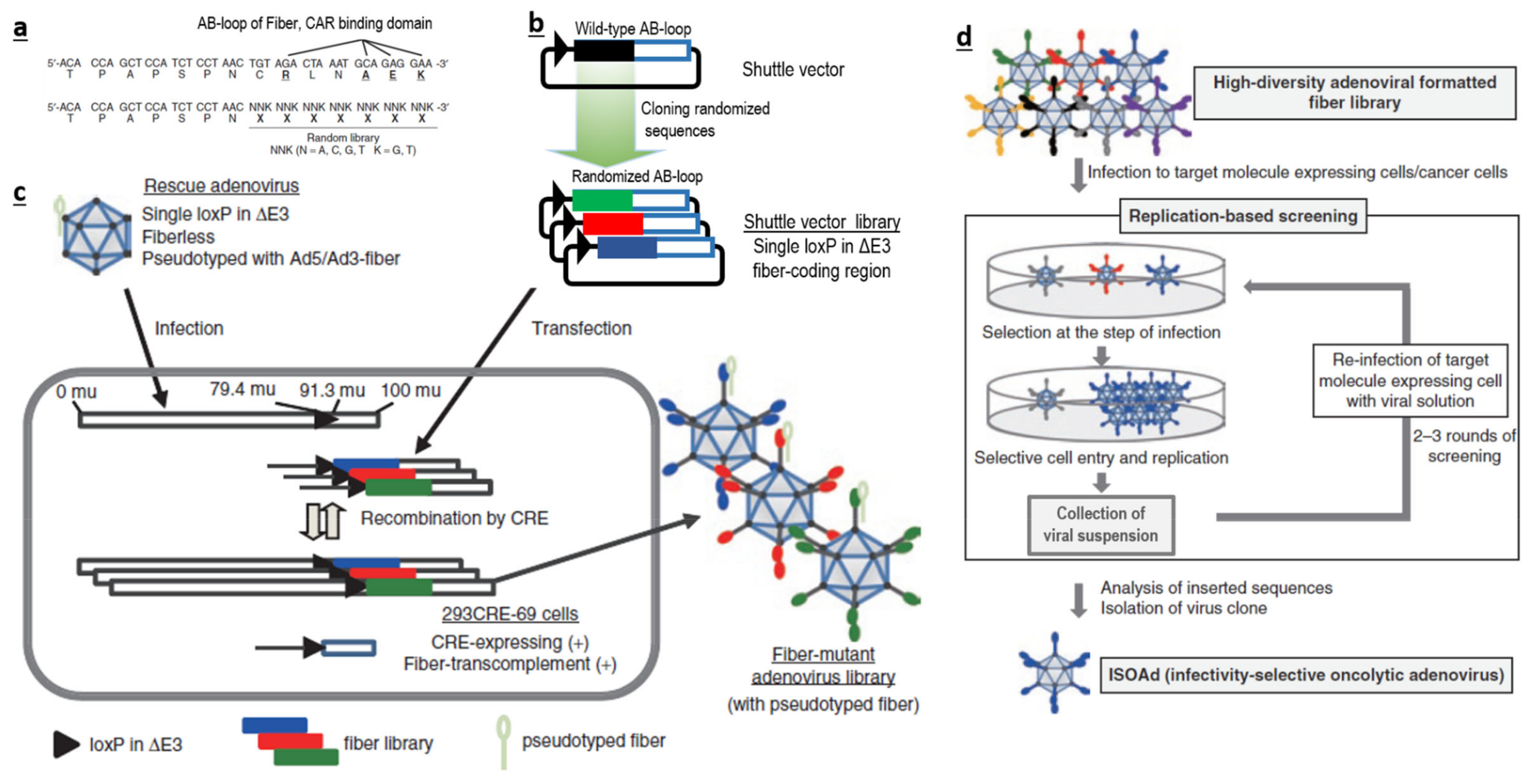
Therefore, if possible, it is more logical to avoid traditional bacterial or chemical screening methods. It could be better to directly generate a large-scale AdV library with huge sequence diversity at the desired loci (Figure 1a–c), and screen them for their ability to interact with a specific target (Figure 1d), instead of using a phage-based system in bacteria. Such AdV-based peptide display libraries can be created in the loops (like AB-loop, HI-loop) of fiber, c-terminal domains of fibers, HVRs of hexon and in pIX. We believe that AB-loop-based libraries are more suitable, as sequence manipulation in this region has a lower chance of affecting capsid assembly and can also abolish interactions between the wild-type AB-loop and CAR. This will consolidate both detargeting and retargeting within one library.
Generating libraries of eukaryotic viruses such as adenoviruses is a more complicated process than phage-based libraries or just plasmid-based libraries. Phage libraries are relatively easier to produce as they require only bacteria, which are more efficient than eukaryotic cells for DNA manipulation and transformation [122]. The difference in efficiency is huge and varies by several magnitudes. Hence, unfortunately, researchers had to choose between screening of a large number of candidates under alien conditions versus a limited number of candidates under native conditions [123][55,123]. Obviously, a library of a large number of candidates that can be screened under native conditions will be an ideal solution.
Several methods have been published, which describe the generation of large libraries, especially of phages and plasmids. Some reports have claimed to achieve libraries with a staggering diversity of over 40 billion unique candidates [124]. However, estimation of diversity in this report was based on statistical extrapolation of fewer than 100 sequences, rather than high-throughput next-generation sequencing (NGS) based methods [124]. Surprisingly, there are several other reports that have used NGS to sequence millions of candidates to more accurately determine the diversity of their libraries [125]. They unambiguously found that such high diversity (40 billion as claimed by others) did not exist in their libraries and it would be very difficult to achieve, even by scaling up their efforts. Very interestingly, in most of the published literature, we found that studies that claim huge diversity have not validated their claims with NGS, while those who used NGS never claim such large numbers. This correlation is revealing and it would be instructive to see the groups claiming huge diversity (billions) use NGS-based methods to validate their libraries and determine the actual diversity. Such studies are still awaited.
Our group has designed a novel approach for generating a library of AdVs with high sequence diversity in the AB-loop regions of fibers [55]. Screenings using this library has led to the identification of two successful candidates against mesothelin and CD133 [55,100]. Further, we have developed novel methods to generate an ultra-high sequence diversity library in the AB-loop region of fiber. The diversity has been validated to contain more than 100 million candidates by using stringent NGS-based methods [126]. This is, by far the largest, validated library (among plasmid, peptide, antibody, phage or recombinant virus based libraries) to our knowledge. Such a library with largest reported diversity which enables screening under native conditions will be very useful tool for screening for novel specificity determinants.
3.1.3 Retargeting of AdV by Affinity Modifiers
Protein- and Peptide-Based Bridging Molecules
High-throughput screening of libraries and validation of selected candidates can be very resource intensive. Conceptually, a simpler way to overcome this, is by using “affinity modifiers” or bridging molecules. Typically, these are capable of binding to a viral particle on one end and to a potential target on the other end. Modifiers can be of a combination of a variety of specificity determinants, including peptide-based linkers, bispecific antibodies, diabodies, triabodies, antibody–ligand complexes, receptor–ligand complexes, and high-affinity binders to virus–ligand complexes.
The main purpose of these modifiers is to bring the virus in close proximity of an intended target cell and facilitate interaction. A few successful demonstrations are listed in Table 1 [127][128][129][130][131][132][133][134][135][127–135].
Table 1. Detargeting and retargeting strategies with representative studies.[136][137][138][139][140][141][142][143][144][145][146][147][148][149][150][151][152][153][154][155][156][157][158][159][160][161][162]
|
Strategy |
Examples |
References |
|
a. Selective Detargeting And Retargeting Towards Specific Receptors/Cells. |
||
|
Detargeting of AdV structural proteins |
||
|
Detargeting in Hexon |
|
|
|
Insertion of lysine in hexon |
Liver detargeting |
[60] |
|
Insertion of targeting peptides |
Skeletal muscle cells, liver detargeting |
[64,65,76–79] |
|
Insertion of large universal acceptor proteins (BAP) |
Liver detargeting |
[80] |
|
HVR swap |
Liver detargeting, breast cancer, immune evasion |
[57,59,61] |
|
Drugs (warfarin, Snake venom protein X-Bp) |
Liver detargeting |
[62,63] |
|
Hexon and pIX |
Liver detargeting, HCC, ovarian carcinoma, melanoma |
[81–83] |
|
Detargeting of Fiber |
|
|
|
Modified HI-loop Modified AB-loop Insertion of proteins or universal acceptors |
neuronal cells, brain endothelial cells, vascular endothelial cells, prostate cancer, lung cancer, pancreatic cancer, muscle cells, colorectal cancer |
[66,67,85–97] [55,100] [87,88,98,99,101–114] |
|
Infectivity Selective Screening for Specific Targeting: Strategies and Importance |
||
|
Phage display and other screening: Insertion of selected peptides into AdV proteins |
Liver detargeting, neuronal cells, brain endothelial cells, vascular endothelial cells, prostate cancer, lung cancer, pancreatic cancer, muscle cells, colorectal cancer |
[79,121] |
|
Adenovirus-library-based screening: Direct modification of AdV libraries. |
Liver detargeting, pancreatic cancer, colorectal cancer. |
[55,100] |
|
Retargeting of AdV by Affinity Modifiers |
||
|
Antibody–antibody-based bridging molecules |
Anti-AdV protein conjugated to VEGFR, TIE-2, integrins, EPCAM, EGFR, HER2, Endoglin, HMWMAA |
[127–132] |
|
Antibody–ligand-based bridging molecules |
anti-AdV protein conjugated to Folate, TNFa, IGF1, EGF |
[133,134] |
|
Peptide–antibody-based bridging molecules |
p75 neurotropin receptor on hepatic stellate cells |
[135] |
|
Soluble receptors conjugated to ligands (sCAR, FX) |
EGF, anti-Cd40 ScFv, ApoE ligand, anti-ErbB2, anti-CEA, Polysialyc-acid (PSA), CXCL12 |
[136–145] |
|
BiTE, Leucine-zipper-based linkers |
CD44v6, anti-B-cell maturation antigen, FR-α, EGFR, EpCAM, carcinoembryonic antigen, CD40 |
[147–154] |
|
b. Selective Expression of Effectors (Inhibitors/Enhancers) in Specific Cells. |
||
|
Protein-based effectors |
Interferons, GM-CSF, IL12, CD40L, CTLA4 |
[156–168,170] |
|
Nucleic-acid-based effectors. |
Liver detargeting, miR122, miR145, miR148, miR21, let7, KRAS, breast cancer, Hepatocellular carcinoma, colorectal cancer cells |
[202–208] |
|
c. Selective replication of virus in specific cells. |
||
|
Promoter-based CRAd |
Lung, prostate, ovarian, pancreatic cancer, adult T cell Leukemia/Lymphoma, glioma, meduloblastoma, sarcomas |
[69,209–222] |
|
CRAds based on Interaction of essential viral genes with Tumor-specific proteins |
colon, breast, non-small cell lung, head and neck, and pancreatic tumors, cervical carcinoma, glioblastoma, cancers with disturbed Rb pathway |
[172,223–228] |
In general, due to the structural complexity of bridging molecules, their production, method of delivery, and stability pose limitations on their application. However, a large number of studies have demonstrated their effectiveness at least in vitro and when no other retargeting methods are available, these inconveniences may be overlooked.
Soluble Receptors Conjugated to Ligands
Curiel’s group reported a novel way of designing bridging molecules by exploiting natural receptors of AdVs. Viruses have naturally evolved high affinity towards specific cell surface receptors. By using a soluble form of the same natural receptor, they could block (saturate) the native viral protein, leading to effective detargeting. This was demonstrated by using a soluble CAR (sCAR) conjugated to several ligands and ScFvs to retarget AdVs [163][164][165][166][167][168][169][170][171][172][136–144]. Hexon– Coagulation Factor (F)X (FX) binding was utilized in a similar manner by conjugating an ScFv against HER2/neu to the FX protein [172][145]. However, the in vivo efficiency of these methods has yet to be demonstrated.
Viruses, which express retargeting molecules as fused structural proteins (e.g., as nanobodies, peptides, or ScFVs fused to viral fiber, penton, pIX, or hexon) may not allow proper folding and post-translational modifications. This may render such retargeting molecules non-functional. This is somewhat expected, as the requirements of maturation of viral proteins and expressed targeting molecules are very different. Hence, expressing these targeting molecules separately and allowing them to undergo full maturation before assembling them into a functional virus–adapter complex could be more effective. However, expressing these retargeting molecules separately and then assembling them with virus particles into a functional complex before delivery has not been easy either. Synthetic leucine-zipper-based dimers [173][174][146,147] and bispecific T-cell engagers (BiTEs) are examples of retargeting molecules that may need extensive maturation. Several studies have tried to use them to increase tumor specificity, however the overall outcomes have not been significant [175][176][177][146,148–155].
3.2. Selective Expression of Effectors (Inhibitors or Enhancers) for Enhanced Specificity
3.2. Selective Expression of Effectors (Inhibitors or Enhancers) for Enhanced Specificity
In addition to selective targeting, selective expression of effectors under spatially and temporally regulated promoters or regulatory elements provides another dimension to specific targeting.
Adenoviruses have been very effective gene delivery vectors, at least in vitro. They can accommodate about 3.5 kb of additional nucleotides in their genome, which is quite reasonable and sufficient to allow insertion of most commonly used gene expression cassettes. Many spatially and temporally regulated promoter driven expression cassettes were engineered into the AdV genome to express essential viral genes and other heterologous genes, including therapeutic proteins (interferons, monoclonal antibodies, cytokines, arresten, TNF-related apoptosis-inducing ligand (TRAIL) [178][179][180][181][182][183][184][185][186][187][188][189][190][191][192][193][194][195][196][156,157,158,159,160,161,162,163,164,165,166,167,168,169], and nucleic-acid-based effectors (RNAi-based regulatory elements, clustered regularly interspaced short palindromic repeats (CRISPR) and, transcription activator-like effector nucleases (TALENs).
3.2.1. Protein-Based Effectors
Hemminki et al. used an OAdV to express granulocyte–macrophage colony-stimulating factor (GMCSF) under a viral E3 promoter (Ad5/3-D24-GMCSF). They were quite efficient in inducing a selective antitumor response. Hemminki et al. and others further demonstrated its effectiveness in patients who had failed conventional chemotherapy and radiation [197][198][156,157,158,159,160,170,171]. Similar to GMCSF, several other receptors and cytokines such as CD40L [162,163,164] and interferons [199][200][201][165,166,167,172,173,174] have also been evaluated. Yamamoto’s group has effectively used interferon expressing OAdVs to target pancreatic cancer in mice models and immunocompetent hamster models [199][200][201][172,173,174]. OAdVs have also been developed to express a monoclonal antibody against CTLA4. Hemminki’s group used a similar Ad5/3-D24aCTLA4 vector to express it in tumor cells and observed selective stimulation of T-cells in patients [195][168]. Most of these effectors are expressed with native adenovirus promoters, however using them with a specific promoter (temporally and/or spatially regulated promoter) may enhance the specificity.
3.2.2. Nucleic-Acid-Based Effectors
Nucleic-acid-based effectors have significant advantages, as they do not need to express any proteins or undergo complicated post-translational modifications (as in the case of RNAi). Even when translation is necessary to produce functional protein–RNA complexes, they do not need to be produced in large quantities, as most of them are multiple turnover catalytic complexes (RNAi, CRISPRs, and TALENs) [202][175].
RNAi is an endogenous pathway of a cell or organism that defends against invasive genetic elements, such as viruses and transposons. RNAi-mediated pathways, which execute their functions through a multitude of small RNA-mediated pathways, including microRNAs, are key to maintaining cellular homeostasis and regulating metabolism [203][204][205][206][176,177,178,179]. Many miRNAs have been found to be involved in tumorigenesis by functioning as oncomiRs and tumor suppressors. Hence, they can also be potential targets for therapy. RNAi as a technique can be used to suppress mis-regulated oncogenes or oncomiRs via siRNAs, shRNAs, artificial miRNAs, anti-miRs, miRzips, sponges, ceRNAs, and artificial lncRNAs [207][208][209][210][177,180,181,182,183]. However, their delivery in vivo has always been a concern [211][177,182,183,184]. Adenoviruses are promising in vivo delivery vectors, at least in some cases [212][213][214][185,186,187] and have the potential to be very useful in this regard.
Similar to many other viruses, AdVs also encode suppressors of RNAi, namely VA1 (viral associated RNA 1) and VA2 RNAs [215][216][217][218][188,189,190,191]. Usually removing such RNAi suppressors is known to enhance the efficiency of RNAi. Machitani et al. demonstrated this by efficiently suppressing a gene, but only with a non-replicating adenovirus [219][192]. On the other hand, the VA (viral associated) RNA is very important for oncolysis. VA RNA being a pro-viral RNA that is essential for efficient virus replication and inhibition of endogenous antiviral pathways, such as the PKR (Protein kinase RNA-activated) pathway, RNAi, and oligo adenylate synthase (OAS) mediated pathways is essential for efficient virus production and infectivity [220][221][222][223][224][193,194,195,196,197]. Removing VA-RNA leads to 20–60-fold decrease in AdV copies, which is very critical for effective oncolytic activity of adenoviruses[224] [197]. Therefore, although the efficiency of the RNAi could be moderate, using replication of competent viruses without removing VA-RNA seems to be more effective for oncolysis [225][226][227][198,199,200,201]. Being very small in size, such RNAi-based effectors could be beneficial in designing combinatorial strategies where the size of the engineered recombinant virus genome is very critical to maintain efficient packaging [228][229][200,201,202].
As with genes, several endogenous miRNAs and other lncRNAs can be targeted to enhance the detargeting and retargeting ability of OAdVs [162][229]. For instance, liver, where most of the OAdV sequestration is observed, overexpresses miRNA122 and miR145. By incorporating multiple copies of miR122/145 target sites (in other words anti-miRs or sponges) in the 3′UTR of essential adenovirus open reading frames ORFs, significant reduction of AdV replication in liver was achieved [136][203]. Many other studies have deployed similar strategies by targeting miR122, miR145, miR148, miR21, and let7 [136][230][231][232][233][234][235][236][237][203,230,231,232,233,234,235,236,237]. These miRNAs, which are overexpressed in liver but not in tumors, are very useful in detargeting of AdV and they can be combined with other strategies. Since they are very small in size, it is a very feasible method.
Similar to genes, which can act as oncogenes or tumor suppressors, miRNAs can also promote (onco-miRs) or inhibit tumorigenesis (tumor suppressors). Typically, onco-miRs can be directly inhibited by using anti-miRs (sponges, ceRNAs) [204,205]. Ang et al. used OAdVs to target endogenous onco-miRs and inhibit EMT and tumor progression in triple-negative breast cancer (TNBC) [204]. They used an artificial lncRNA consisting of targets of nine onco-miRs overexpressed in TNBC cells, to suppress them simultaneously. In another study, an artificial lncRNA was used to suppress six different miRNAs simultaneously in sorafenib-resistant hepatocellular carcinomas (HCCs) [205]. These cancers are usually difficult to treat and this study showed the potential of RNAi-based therapies. These strategies are very versatile, as they can be used to target multiple genes or miRNAs together, which otherwise is very difficult to accomplish [238].
Sometimes, overexpression of miRNAs (tumor suppressor miRNAs) could be beneficial in reducing tumor burden by suppressing overexpressed oncogenes. For example, miR143 was overexpressed using OAdVs to suppress an oncogene, Kirsten rat sarcoma viral oncogene homolog (KRAS), in colorectal cancer cells [206] and miR199 was overexpressed to address HCC [203].
In absence of natural miRNAs to suppress oncogenes, artificial miRNAs, siRNAs, and shRNAs can be used [182,183,203,206,207]. The FGL2 gene in HCC tumors was targeted by using such artificial miRNAs to suppress angiogenesis [207].
Therefore, miRNAs, depending on their role, can be exploited for detargeting, retargeting, and as therapeutic agents by OAdVs. Combining them with other strategies will enhance the potency of OAdV-based therapies [123,124]. Luo et al. used a triple-regulated OAdV carrying miR143, survivin, and RGD to enhance the effects of OAdVs [124]. These RNAi methods are very versatile as they can be used to target multiple genes or miRNAs simultaneously, which is difficult using any other method. Since their size is relatively small, they can be easily engineered into some of the well-established and commonly used vectors [200,201,202].
Unlike RNAi, which is mostly a post-transcriptional gene silencing mechanism, TALENs and CRISPRs have revolutionized genome engineering and have opened up new avenues of targeted therapy [239][240][241][243][175,239,240,241]. Oncogenes and tumor suppressors can be manipulated using these techniques [241][242][241,242]. Again, their delivery has been a big challenge. In some cases, adenoviruses were effectively used to deliver these programmable nucleases or modifiers [244][245][246][247][243,244,245,246,247]. Among these genome engineering techniques, the nucleotide components determining the sequence specificity in CRISPRs (guides) are smaller than those of TALENs. Hence, the CRISPR-based strategies seem more promising than TALENs. They can potentially be accommodated in a non-helper-dependent viral vector, although most of the studies so far have used helper-dependent viruses [248][249][250][251][252][175,246,248,249,250,251,252]. Maggio et al. delivered functional gRNAs and human-codon-optimized Cas-9 into a diverse array of human cells [244]. Although some gene editing-based studies have shown promising results and are going through clinical trials [253][254][253,254], creating mutations in the genome may not be a safe strategy, due to its permanence and potential off-target activities. This may impart undesirable and irreversible damages to genome. However, other CRIPR based tools like, modified CRISPR-based base editors, suppressors and enhancers (which do not cleave the target) could be safer alternatives [255][256][257][258][248,249,250,255,256,257,258].
3.3. Selective (Conditional) Replication of Viruses in Specific Cells
The ability of oncolytic adenoviruses to replicate specifically in cancer cells will provide them a significant advantage over traditional chemotherapies. In order to achieve this, an oncolytic adenovirus must: (i) demonstrate stability in vivo and avoid potential degradation or uptake by non-target cells; and (ii) preferentially infect and replicate in cancer cells, thereby providing limited delivery to normal cells via a conditionally replicative adenovirus (CRAd) [259]. There are mainly two types of CRAds: one uses a tumor-specific promoter to induce replication and the second uses the interaction of essential viral genes with tumor-specific proteins.
Several genes are uniquely expressed or significantly overexpressed in tumors, often due to their promoters being active in those tumor cells and microenvironments. Promoter-based CRAds are designed to express essential viral genes with such promoters so that virus replication is possible only in those cells. The most prominent examples include Cox2 in gastric cancer and pancreatic cancer [69,209,210,211] and survivin in adult T cell leukemia or lymphoma [212,213,214,215] and lung, ovarian, and pancreatic cancers. The HIF-responsive promoter in gliomas and meduloblastomas [216,217]; and hTERT (human telomerase reverse transcriptase) in bone and soft tissue sarcomas, as well as prostate, ovarian, esophageal, and GI cancers [260][261][218,219,220,221,222,260,261], among many others.
The second type of CRAds involve mutations or deletions in the E1 region to allow tumor-specific replication [262][223,262]. The E1 region of the adenovirus is involved in its replication and consist of 2 genes, E1A and E1B.These two genes encode proteins essential for a “productive” adenovirus replication cycle [263]. E1A encodes for proteins 243 R and 289 R, which induce transcription of early viral gene regions and stimulate the entry of infected cells to the S phase of the cell cycle. E1B encodes for proteins E1B19K and E1B55K, which are involved in inhibition of apoptosis via their interaction with p53 and Rb proteins in infected cells, among other important functions [263]. E1A causes the induction of the S phase, which has an apoptotic response and must be inhibited by E1B gene products in order to facilitate successful viral replication cycle [264][265][266][264]. Mutations or deletions in the E1 region abolish the interaction of viral proteins with either p53 via E1B [265][266][265,266] or pRb via E1A [267][268][267,268] to target tumor cells defective for those gene products. ONYX-015 is a OAdV that lacks the EB1B region and selectively replicates in mutated p53 tumors [224]. Similarly, Ad∆24 is a OAd that has a mutation in E1A and restricts replication to retinoblastoma protein (pRb) mutated cancer cells [225]. Although these CRAds are better than first-generation CRAds, such as onyx-015, they were not very effective in clinical trials when used alone [269]. To target cancer cells that harbor activating mutant KRAS (KRASaMut) but spare p53wild normal cells, Liu et al. [270] constructed the Δp53REP2 promoter with deletion of its p53-response elements. This was used to regulate the expression of the hdm2 transgene in a novel E1B-55kD-deleted CRAd, the Ad-KRhdm2. The virus showed selective replication in colorectal cancer cells with KRAS mutation and P53 WT. Furthermore, Kim et al., in an effort to augment radiation therapy, generated double E1B 19kDa and E1B 55kDa deleted oncolytic adenovirus (Ad−ΔE1B55), which when combined with radiation therapy showed greater cytotoxicity than the single E1B-55kDA-deleted oncolytic AdV [271]. Similarly, Yoon et al. [272] demonstrated that combined deletion of E1B 19kDA and E1B 55kDA increased the cytotoxicity when combined with cisplatin, which is a standard of care for many cancers, such as ovarian, cervical, breast, and bladder cancers.
3.4. Combinations of the Abovementioned Strategies and Other Anticancer Treatments for Enhanced Specificity and More Effective Therapeutic Effect
We believe a combination of detargeting and retargeting strategies is more effective than either of them alone. This has been demonstrated by combinations of targeted modifications in the host-interacting domains of viral proteins, such as HVR regions of hexon and AB-loop regions of fiber [55,78,100].
Since a replication-competent adenovirus genome can accommodate up to ~3.5 kb of extra genetic material into its capsid, other strategies to increase the specificity and efficiency can be combined. Selective expression of effectors and conditional replication-based strategies are best combined with detargeting- and retargeting-based vector designs.
Nucleic-acid-based effectors (RNAi-based) are smaller in size and relatively easier to combine with other strategies. Therefore, miRNAs, depending on their role, can be exploited for detargeting, retargeting and also as therapeutic agents by OAdVs [202,208]. Luo et al. used a triple-regulated OAdV carrying miR143, survivin, and RGD to enhance the effects of OAdVs [200,202]. Lou et al. overexpressed miR34a combined with overexpression of interleukin-24 (IL-24) using hTERT promoter-driven E1A-D24-type CRAds to test against HCC [208]. These methods are very versatile, as they can be used to target multiple genes or miRNAs simultaneously, which is difficult to do using any other method. Some studies have targeted up to nine miRNAs at once [205]. Since their size is small they can be easily engineered into well established and commonly used vectors [200].
Protein-based effectors such as interferon-α show toxicity when administered systemically [273]. Yamamoto’s group developed an OAdV (Ad5/Ad3-Cox2-ΔE3-ADP-IFN) that combines multiple strategies, including conditional replication, detargeting and retargeting via the RGD motif and Ad5/3 chimeric fiber, overexpression of ADP and expression of interferon-α to target pancreatic cancer [173,174]. Another OAdV (CRAd-arresten-TRAIL) was developed to combine conditional replication, engineered fibers, and expression of two protein effectors, arresten and TRAIL [169]. Hemmiki’s group used OAdVs (Ad5/3-Δ24aCTLA4) to express checkpoint inhibitors such as anti-CTLA4 in combination with Rb/p16-dependent CRAd and chimeric fiber [167] to observe enhanced stimulation of T-cells in patients with advanced cancer.
Combining these strategies to improve tumor selectivity and specificity with other anticancer treatments, such as chemotherapy and radiotherapy, can further improve the therapeutic outcomes [274]. An interferon-expressing OAdV used in combination with chemotherapy and radiotherapy significantly reduced the systemic toxicity and increased antitumor effects [172]. OAdVs seem to make targeted cells more sensitive to radiation-induced damages by suppressing dsDNA break repair pathways [275][276][277][226,275,276,277]. Their interaction with other conventional chemotherapies are not clear but they have been found to be effective, as shown in the case of onyx-015 and other interferon-expressing CRAds [276][277][172,226,227]. In some cases, specific inhibitors of oncogenes were combined with CRAds. Nutins, known inhibitors of MDM2, which in turn suppresses p53, were used in combination with a CRAd designed to overexpress p53 to get a much better effect than with p53-overexpressing AdV alone [278][228].
Overall, combinations of these strategies enhance tumor selectivity and specificity. Depending on the context, such as the type of tumor, genes needed to be targeted, and other conventional therapies available, one can tailor multiple combinatorial strategies for a better therapeutic outcome.
4. Conclusions
- Conclusions
Oncolytic viruses are novel and effective tools for delivering targeted therapy to cancer cells. They have the potential to target any gene that is specifically expressed or overexpressed on the surface of a cancer cell. Most importantly, OVs can exploit genes which are overexpressed, irrespective of their functional relevance to tumor biology. This makes a large number of genes targetable which could not be targeted earlier by conventional therapies. The flexibility of DNA manipulation to drive detargeting and retargeting of these viruses, combined with conditional replication and targeted expression, allows for the combination of multiple good tools into one. This may enable us to design a therapy that is more specific, targeted, and effective. Furthermore, the combination of these new therapies with conventional anticancer therapies such as radiation and chemotherapy is very promising and may provide additional benefits to cancer patients
References
- Henley, S.J.; Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.-M.; Howlader, N.; Anderson, R.N.; Firth, A.U.; Ma, J.A. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer 2020, 126, 2225–2249.
- Henley, S.J.; Thomas, C.C.; Lewis, D.R.; Ward, E.M.; Islami, F.; Wu, M.; Weir, H.K.; Scott, S.; Sherman, R.L.; Ma, J. Annual report to the nation on the status of cancer, part II: Progress toward Healthy People 2020 objectives for 4 common cancers. Cancer 2020, 126, 2250–2266.
- Davies, A.; Lum, C.; Raju, R.; Ansell, E.; Webber, K.; Segelov, E. Anti-cancer therapy made easier: A 25-year update. Intern. Med. J. 2020, doi:10.1111/imj.14878.Davies, A.; Lum, C.; Raju, R.; Ansell, E.; Webber, K.; Segelov, E. Anti-cancer therapy made easier: A 25-year update. Intern. Med. J. 2020.
- Rizvi, S.A.A.; Shahzad, Y.; Saleh, A.M.; Muhammad, N. Dose Issues in Cancer Chemotherapy. Oncology 2020, 1–8, doi:10.1159/000506705.Rizvi, S.A.A.; Shahzad, Y.; Saleh, A.M.; Muhammad, N. Dose Issues in Cancer Chemotherapy. Oncology 2020, 1–8.
- Ke, X.; Shen, L. Molecular targeted therapy of cancer: The progress and future prospect. Front. Lab. Med. 2017, 1, 69–75.
- Haley, B.; Roudnicky, F. Functional Genomics for Cancer Drug Target Discovery. Cancer Cell 2020, doi:10.1016/j.ccell.2020.04.006.Haley, B.; Roudnicky, F. Functional Genomics for Cancer Drug Target Discovery. Cancer Cell 2020.
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196.
- Alessandrini, L.; Perin, T.; Kadare, S.; del Pup, L.; Memeo, L.; Steffan, A.; Colarossi, L.; Berretta, M.; de Paoli, P.; Canzonieri, V. Cancer Targeted Therapy Strategy: The Pathologist’s Perspectives. Curr. Cancer Drug Targets 2018, 18, 410–420.
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519.
- Eder, J.; Sedrani, R.; Wiesmann, C. The discovery of first-in-class drugs: Origins and evolution. Nat. Rev. Drug Discov. 2014, 13, 577–587.
- Roy, A. Early Probe and Drug Discovery in Academia: A Minireview. High Throughput 2018, 7, 4.Roy, A. Early Probe and Drug Discovery in Academia: A Mini Review. High Throughput 2018, 7, 4.
- Von Stechow, L.; Cancer Systems Biology: Methods and Protocols; Springer: New York, NY, USA, 2018.Von Stechow, L. Cancer Systems Biology: Methods and Protocols; Springer: New York, NY, USA, 2018.
- DeWard, A.; Critchley-Thorne, R.J. Systems Biology Approaches in Cancer Pathology. Methods Mol. Biol. 2018, 1711, 261–273.
- Kibble, M.; Saarinen, N.; Tang, J.; Wennerberg, K.; Mäkelä, S.; Aittokallio, T. Network pharmacology applications to map the unexplored target space and therapeutic potential of natural products. Nat. Prod. Rep. 2015, 32, 1249–66.Kibble, M.; Saarinen, N.; Tang, J.; Wennerberg, K.; Mäkelä, S.; Aittokallio, T. Network pharmacology applications to map the unexplored target space and therapeutic potential of natural products. Nat. Prod. Rep. 2015, 32, 1249–1266.
- Barneh, F.; Jafari, M.; Mirzaie, M. Updates on drug-target network; facilitating polypharmacology and data integration by growth of DrugBank database. Brief Bioinform.2016, 17, 1070–1080.Barneh, F.; Jafari, M.; Mirzaie, M. Updates on drug-target network; facilitating polypharmacology and data integration by growth of DrugBank database. Brief Bioinform. 2016, 17, 1070–1080.
- Smith, R.D.; Hu, L.; Falkner, J.A.; Benson, M.L.; Nerothin, J.P.; Carlson, H.A. Exploring protein-ligand recognition with Binding MOAD. J. Mol. Graph. Model 2006, 24, 414–425, doi:10.1016/j.jmgm.2005.08.002.Smith, R.D.; Hu, L.; Falkner, J.A.; Benson, M.L.; Nerothin, J.P.; Carlson, H.A. Exploring protein-ligand recognition with Binding MOAD. J. Mol. Graph. Model 2006, 24, 414–425.
- Coussens, N.P.; Braisted, J.C.; Peryea, T.; Sittampalam, G.S.; Simeonov, A.; Hall, M.D. Small-Molecule Screens: A Gateway to Cancer Therapeutic Agents with Case Studies of Food and Drug Administration-Approved Drugs. Pharmacol. Rev. 2017, 69, 479–496.
- Couto, G.K.; Segatto, V.N.; Oliveira, L.T.; Seixas, K.F.; Schachtschneider, M.K.; Collares, T. The Melding of Drug Screening Platforms for Melanoma. Front. Oncol. 2019, 9, 512.
- Ma, M.; Liu, J.; Jin, S.; Wang, L. Development of tumour peptide vaccines: From universalization to personalization. Scand. J. Immunol. 2020, 91, e12875.
- Saw, P.E.; Song, E.W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807.
- Parakh, S.; King, D.; Gan, H.K.; Scott, A.M. Current Development of Monoclonal Antibodies in Cancer Therapy. Recent Results Cancer Res. 2020, 214, 1–70.
- Ma, X.; Lakshmipriya, T.; Gopinath, S.C.B. Recent Advances in Identifying Biomarkers and High-Affinity Aptamers for Gynecologic Cancers Diagnosis and Therapy. J. Anal. Methods Chem. 2019, 2019, 5426974.
- Kogej, T.; Blomberg, N.; Greasley, P.J.; Mundt, S.; Vainio, M.J.; Schamberger, J.; Schmidt, G.; Hüser, J. Big pharma screening collections: More of the same or unique libraries? The AstraZeneca-Bayer Pharma AG case. Drug Discov. Today 2013, 18, 1014–1024.
- Chai, C.L.; Matyus, P. One size does not fit all: Challenging some dogmas and taboos in drug discovery. Future Med. Chem. 2016, 8, 29–38.
- Posner, B.A. High-throughput screening-driven lead discovery: Meeting the challenges of finding new therapeutics. Curr. Opin. Drug Discov. Devel. 2005, 8, 487–494.
- Kinast, V.; Burkard, T.L.; Todt, D.; Steinmann, E. Hepatitis E Virus Drug Development. Viruses 2019, 11, 485.
- Davola, M.E.; Mossman, K.L. Oncolytic viruses: How “lytic” must they be for therapeutic efficacy? Oncoimmunology 2019, 8, e1581528.
- Allan, K.J.; Stojdl, D.F.; Swift, S.L. High-throughput screening to enhance oncolytic virus immunotherapy. Oncolytic Virother. 2016, 5, 15–25.
- Ring, C.J.A. Cytolytic viruses as potential anti-cancer agents. J. Gen. Virol. 2002, 83, 491–502.
- Hermiston, T.W.; Kuhn, I. Armed therapeutic viruses: Strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002,9 1022-35.Hermiston, T.W.; Kuhn, I. Armed therapeutic viruses: Strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002, 9, 1022–1035.
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662.
- Davydova, J.; Yamamoto, M. Oncolytic adenoviruses: Design, generation, and experimental procedures. Curr. Protoc. Hum. Genet. 2013, 78, 12.14.1–12.14.2.Davydova, J.; Yamamoto, M. Oncolytic adenoviruses: Design, generation, and experimental procedures. Curr. Protoc. Hum. Genet. 2013, 78, 12–14.
- Russell, L.; Peng, K.W. The emerging role of oncolytic virus therapy against cancer. Chin. Clin. Oncol. 2018, 7, 16.
- Ahmad, T.; Venkataraman, S.; AbouHaidar, M.; Hefferon, K.L. Recent Patents in Oncolytic Virotherapy. Recent Pat. Biotechnol. 2016, 9, 79–85.
- Niemann, J.; Kuhnel, F. Oncolytic viruses: Adenoviruses. Virus Genes 2017, 53, 700–706.
- Bishnoi, S.; Tiwari, R.; Gupta, S.; Byrareddy, S.N.; Nayak, D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy. Viruses 2018, 10, 90.
- Watanabe, D.; Goshima, F. Oncolytic Virotherapy by HSV. Adv. Exp. Med. Biol. 2018, 1045, 63–84.
- Lauer, U.M.; Schell, M.; Beil, J.; Berchtold, S.; Koppenhöfer, U.; Glatzle, J.; Königsrainer, A.; Möhle, R.; Nann, D.; Fend, F. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clin. Cancer Res.2018, 24, 4388–4398.Lauer, U.M.; Schell, M.; Beil, J.; Berchtold, S.; Koppenhöfer, U.; Glatzle, J.; Königsrainer, A.; Möhle, R.; Nann, D.; Fend, F. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clin. Cancer Res. 2018, 24, 4388–4398.
- Kemp, V.; Hoeben, R.C.; van den Wollenberg, D.J. Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses 2015, 8, 4, doi:10.3390/v8010004.Kemp, V.; Hoeben, R.C.; van den Wollenberg, D.J. Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses 2015, 8, 4.
- Zhao, X.; Cariad, C.; Rajasekaran, N.; He, Z.X.; Kohrt, H.E. Strategic Combinations: The Future of Oncolytic Virotherapy with Reovirus. Mol. Cancer 2016, 15, 767–773.
- Zhao, X.; Cariad Chester, Rajasekaran, N.; He, Z.X.; Kohrt, H.E. Strategic Combinations: The Future of Oncolytic Virotherapy with Reovirus. Mol. Cancer 2016, 15, 767–773.Burke, M.J. Oncolytic Seneca Valley Virus: Past perspectives and future directions. Oncolytic Virother. 2016, 5, 81–89.
- Burke, M.J. Oncolytic Seneca Valley Virus: Past perspectives and future directions. Oncolytic Virother. 2016, 5, 81–89.Reddy, P.S.; Burroughs, K.D.; Hales, L.M.; Ganesh, S.; Jones, B.H.; Idamakanti, N.; Hay, C.; Li, S.S.; Skele, K.L.; Vasko, A.-J. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J. Natl. Cancer Inst. 2007, 99, 1623–1633.
- Reddy, P.S.; Burroughs, K.D.; Hales, L.M.; Ganesh, S.; Jones, B.H.; Idamakanti, N.; Hay, C.; Li, S.S.; Skele, K.L.; Vasko, A.-J. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J. Natl. Cancer Inst. 2007, 99, 1623–1633.Curiel, D.T. Adenoviral Vectors for Gene Therapy; Elsevier Science: Cambridge, MA, USA, 2016.
- Curiel, D.T. Adenoviral Vectors for Gene Therapy; Elsevier Science: Cambridge, MA, USA, 2016.Kozarsky, K.F.; Wilson, J.M. Gene therapy: Adenovirus vectors. Curr. Opin. Genet. Dev. 1993, 3, 499–503.
- Kozarsky, K.F.; Wilson, J.M. Gene therapy: Adenovirus vectors. Curr. Opin. Genet. Dev. 1993, 3, 499–503.Mitani, K.; Kubo, S. Adenovirus as an integrating vector. Curr. Gene 2002, 2, 135–144.
- Mitani, K.; Kubo, S. Adenovirus as an integrating vector. Curr. Gene 2002, 2, 135–144.Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Arenas, S.S.; Sirima, S.B.; Dzomo, G.R.T.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K. A review of 65 years of human adenovirus seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613.
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Arenas, S.S.; Sirima, S.B.; Dzomo, G.R.T.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K. A review of 65 years of human adenovirus seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613.Seiradake, E.; Henaff, D.; Wodrich, H.; Billet, O.; Perreau, M.; Hippert, C.; Mennechet, F.; Schoehn, G.; Lortat-Jacob, H.; Dreja, H. The cell adhesion molecule “CAR” and sialic acid on human erythrocytes influence adenovirus in vivo biodistribution. PLoS Pathog. 2009, 5, e1000277.
- Seiradake, E.; Henaff, D.; Wodrich, H.; Billet, O.; Perreau, M.; Hippert, C.; Mennechet, F.; Schoehn, G.; Lortat-Jacob, H.; Dreja, H. The cell adhesion molecule “CAR” and sialic acid on human erythrocytes influence adenovirus in vivo biodistribution. PLoS Pathog. 2009, 5, e1000277.Dechecchi, M.C.; Melotti, P.; Bonizzato, A.; Santacatterina, M.; Chilosi, M.; Cabrini, G. Heparan sulfate glycosaminoglycans are receptors sufficient to mediate the initial binding of adenovirus types 2 and 5. J. Virol. 2001, 75, 8772–8780.
- Dechecchi, M.C.; Melotti, P.; Bonizzato, A.; Santacatterina, M.; Chilosi, M.; Cabrini, G. Heparan sulfate glycosaminoglycans are receptors sufficient to mediate the initial binding of adenovirus types 2 and 5. J. Virol. 2001, 75, 8772–8780.Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.K.; Greig, J.A.; Denby, L. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409.
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.K.; Greig, J.A.; Denby, L. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409.Chaurasiya, S.; Chen, N.G.; Fong, Y. Oncolytic viruses and immunity. Curr. Opin. Immunol. 2018, 51, 83–90.
- Chaurasiya, S.; Chen, N.G.; Fong, Y. Oncolytic viruses and immunity. Curr. Opin. Immunol. 2018, 51, 83–90.Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158.
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, Gu.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158.Stone, D.; Liu, Y.; Shayakhmetov, D.; Li, Z.; Ni, S.; Lieber, A. Adenovirus-platelet interaction in blood causes virus sequestration to the reticuloendothelial system of the liver. J. Virol. 2007, 81, 4866–4871.
- Stone, D.; Liu, Y.; Shayakhmetov, D.; Li, Zo.; Ni, S.; Lieber, A. Adenovirus-platelet interaction in blood causes virus sequestration to the reticuloendothelial system of the liver. J. Virol. 2007, 81, 4866–4871.Rojas, L.A.; Moreno, R.; Calderón, H.; Alemany, R. Adenovirus coxsackie adenovirus receptor-mediated binding to human erythrocytes does not preclude systemic transduction. Cancer Gene 2016, 23, 411–414.
- Rojas, L.A.; Moreno, R.; Calderón, H.; Alemany, R. Adenovirus coxsackie adenovirus receptor-mediated binding to human erythrocytes does not preclude systemic transduction. Cancer Gene 2016, 23, 411–414.Carlisle, R.C.; Di, Y.; Cerny, A.M.; Sonnen, A.F.; Sim, R.B.; Green, N.K.; Subr, V.; Ulbrich, K.; Gilbert, R.J.C.; Fisher, K.D. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood 2009, 113, 1909–1918.
- Carlisle, R.C.; Di, Y.; Cerny, A.M.; Sonnen, A.F.; Sim, R.B.; Green, N.K.; Subr, V.; Ulbrich, K.; Gilbert, R.J.C.; Fisher, K.D. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood 2009, 113, 1909–1918.Miura, Y.; Yamasaki, S.; Davydova, J.; Brown, E.; Aoki, K.; Vickers, S.; Yamamoto, M. Infectivity-selective oncolytic adenovirus developed by high-throughput screening of adenovirus-formatted library. Mol. Ther. 2013, 21, 139–148.
- Miura, Y.; Yamasaki, S.; Davydova, J.; Brown, E.; Aoki, K.; Vickers, S.; Yamamoto, M. Infectivity-selective oncolytic adenovirus developed by high-throughput screening of adenovirus-formatted library. Mol. Ther. 2013, 21, 139–148.Hulin-Curtis, S.L.; Uusi-Kerttula, H.; Jones, R.; Hanna, L.; Chester, J.D.; Parker, A.L. Evaluation of CD46 re-targeted adenoviral vectors for clinical ovarian cancer intraperitoneal therapy. Cancer Gene 2016, 23, 229–234.
- Hulin-Curtis, S.L.; Uusi-Kerttula, H.; Jones, R.; Hanna, L.; Chester, J.D.; Parker, A.L. Evaluation of CD46 re-targeted adenoviral vectors for clinical ovarian cancer intraperitoneal therapy. Cancer Gene 2016, 23, 229–234.Roberts, D.M.; Nanda, A.; Havenga, M.J.E.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243.
- Roberts, D.M.; Nanda, A.; Havenga, M.J.E.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243.Khare, R.; Reddy, V.S.; Nemerow, G.R.; Barry, M.A. Identification of adenovirus serotype 5 hexon regions that interact with scavenger receptors. J. Virol. 2012, 86, 2293–2301.
- Khare, R.; Reddy, V.S.; Nemerow, G.R.; Barry, M.A. Identification of adenovirus serotype 5 hexon regions that interact with scavenger receptors. J. Virol. 2012, 86, 2293–2301.Khare, R.; May, S.M.; Vetrini, F.; Weaver, E.A.; Palmer, D.; Rosewell, A.; Grove, N.; Ng, P.; Barry, M.A. Generation of a Kupffer cell-evading adenovirus for systemic and liver-directed gene transfer. Mol. Ther. 2011, 19, 1254–1262.
- Khare, R.; May, S.M.; Vetrini, F.; Weaver, E.A.; Palmer, D.; Rosewell, A.; Grove, N.; Ng, P.; Barry, M.A. Generation of a Kupffer cell-evading adenovirus for systemic and liver-directed gene transfer. Mol. Ther. 2011, 19, 1254–1262.Prill, J.M.; Espenlaub, S.; Samen, U.; Engler, T.; Schmidt, E.; Vetrini, F.; Rosewell, A.; Grove, N.; Palmer, D.; Ng, P. Modifications of adenovirus hexon allow for either hepatocyte detargeting or targeting with potential evasion from Kupffer cells. Mol. Ther. 2011, 19, 83–92.
- Prill, J.M.; Espenlaub, S.; Samen, U.; Engler, T.; Schmidt, E.; Vetrini, F.; Rosewell, A.; Grove, N.; Palmer, D.; Ng, P. Modifications of adenovirus hexon allow for either hepatocyte detargeting or targeting with potential evasion from Kupffer cells. Mol. Ther. 2011, 19, 83–92.Zhang, Z.; Krimmel, J.; Zhang, Z.; Hu, Z.; Seth, P. Systemic delivery of a novel liver-detargeted oncolytic adenovirus causes reduced liver toxicity but maintains the antitumor response in a breast cancer bone metastasis model. Hum. Gene. Ther. 2011, 22, 1137–1142.
- Zhang, Z.; Krimmel, J.; Zhang, Z.; Hu, Z.; Seth, P. Systemic delivery of a novel liver-detargeted oncolytic adenovirus causes reduced liver toxicity but maintains the antitumor response in a breast cancer bone metastasis model. Hum. Gene. Ther. 2011, 22, 1137–1142.Parker, A.L.; Waddington, S.N.; Nicol, C.G.; Shayakhmetov, D.M.; Buckley, S.M.; Denby, L.; Kemball-Cook, G.; Ni, S.; Lieber, A.; McVey, J.H. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 2006, 108, 2554–2561.
- Parker, A.L.; Waddington, S.N.; Nicol, C.G.; Shayakhmetov, D.M.; Buckley, S.M.; Denby, L.; Kemball-Cook, G.; Ni, S.; Lieber, A.; McVey, J.H. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 2006, 108, 2554–2561.Atoda, H.; Ishikawa, M.; Mizuno, H.; Morita, T. Coagulation factor X-binding protein from Deinagkistrodon acutus venom is a Gla domain-binding protein. Biochemistry 1998, 37, 17361–17370.
- Atoda, H.; Ishikawa, M.; Mizuno, H.; Morita, T. Coagulation factor X-binding protein from Deinagkistrodon acutus venom is a Gla domain-binding protein. Biochemistry 1998, 37, 17361–17370.Stepanenko, A.A.; Chekhonin, V.P. Tropism and transduction of oncolytic adenovirus 5 vectors in cancer therapy: Focus on fiber chimerism and mosaicism, hexon and pIX. Virus Res. 2018, 257, 40–51.
- Stepanenko, A.A.; Chekhonin, V.P. Tropism and transduction of oncolytic adenovirus 5 vectors in cancer therapy: Focus on fiber chimerism and mosaicism, hexon and pIX. Virus Res. 2018, 257, 40–51.Vigne, E.; Mahfouz, I.; Dedieu, J.F.; Brie, A.; Perricaudet, M.; Yeh, P. RGD inclusion in the hexon monomer provides adenovirus type 5-based vectors with a fiber knob-independent pathway for infection. J. Virol. 1999, 73, 5156–5161.
- Vigne, E.; Mahfouz, I.; Dedieu, J.F.; Brie, A.; Perricaudet, M.; Yeh, P. RGD inclusion in the hexon monomer provides adenovirus type 5-based vectors with a fiber knob-independent pathway for infection. J. Virol. 1999, 73, 5156–5161.Wu, H.; Seki, T.; Dmitriev, I.; Uil, T.; Kashentseva, E.; Han, T.; Curiel, D.T. Double modification of adenovirus fiber with RGD and polylysine motifs improves coxsackievirus-adenovirus receptor-independent gene transfer efficiency. Hum. Gene Ther. 2002, 13, 1647–1653.
- Wu, H.; Seki, T.; Dmitriev, I.; Uil, T.; Kashentseva, E.; Han, T.; Curiel, D.T. Double modification of adenovirus fiber with RGD and polylysine motifs improves coxsackievirus-adenovirus receptor-independent gene transfer efficiency. Hum. Gene Ther. 2002, 13, 1647–1653.Majhen, D.; Richardson, J.; Vukelić, B.; Dodig, I.; Cindrić, M.; Benihoud, K.; Ambriović-Ristov, A. The disulfide bond of an RGD4C motif inserted within the Hi loop of the adenovirus type 5 fiber protein is critical for retargeting to alphav -integrins. J. Gene Med. 2012, 14, 788–797.
- Majhen, D.; Richardson, J.; Vukelić, B.; Dodig, I.; Cindrić, M.; Benihoud, K.; Ambriović-Ristov, A. The disulfide bond of an RGD4C motif inserted within the Hi loop of the adenovirus type 5 fiber protein is critical for retargeting to alphav -integrins. J. Gene Med. 2012, 14, 788–797.Volk, A.L.; Rivera, A.A.; Kanerva, A.; Bauerschmitz, G.; Dmitriev, I.; Nettelbeck, D.M.; Curiel, D.T. Enhanced adenovirus infection of melanoma cells by fiber-modification: Incorporation of RGD peptide or Ad5/3 chimerism. Cancer Biol. Ther. 2003, 2, 511–515.
- Volk, A.L.; Rivera, A.A.; Kanerva, A.; Bauerschmitz, G.; Dmitriev, I.; Nettelbeck, D.M.; Curiel, D.T. Enhanced adenovirus infection of melanoma cells by fiber-modification: Incorporation of RGD peptide or Ad5/3 chimerism. Cancer Biol. Ther. 2003, 2, 511–515.Davydova, J.; Le, L.P.; Gavrikova, T.; Wang, M.; Krasnykh, V.; Yamamoto, M. Infectivity-enhanced cyclooxygenase-2-based conditionally replicative adenoviruses for esophageal adenocarcinoma treatment. Cancer Res. 2004, 64, 4319–4327.
- Davydova, J.; Le, L.P.; Gavrikova, T.; Wang, M.; Krasnykh, V.; Yamamoto, M. Infectivity-enhanced cyclooxygenase-2-based conditionally replicative adenoviruses for esophageal adenocarcinoma treatment. Cancer Res. 2004, 64, 4319–4327.Gamble, L.J.; Ugai, H.; Wang, M.; Borovjagin, A.V.; Matthews, Q.L. Therapeutic efficacy of an oncolytic adenovirus containing RGD ligand in minor capsid protein IX and Fiber, Delta24DoubleRGD, in an ovarian cancer model. J. Mol. Biochem. 2012, 1, 26–39.
- Gamble, L.J.; Ugai, H.; Wang, M.; Borovjagin, A.V.; Matthews, Q.L. Therapeutic efficacy of an oncolytic adenovirus containing RGD ligand in minor capsid protein IX and Fiber, Delta24DoubleRGD, in an ovarian cancer model. J. Mol. Biochem. 2012, 1, 26–39.Rojas, J.J.; Gimenez-Alejandre, M.; Gil-Hoyos, R.; Cascallo, M.; Alemany, R. Improved systemic antitumor therapy with oncolytic adenoviruses by replacing the fiber shaft HSG-binding domain with RGD. Gene Ther. 2012, 19, 453–457.
- Rojas, J.J.; Gimenez-Alejandre, M.; Gil-Hoyos, R.; Cascallo, M.; Alemany, R. Improved systemic antitumor therapy with oncolytic adenoviruses by replacing the fiber shaft HSG-binding domain with RGD. Gene Ther. 2012, 19, 453–457.Tyler, M.A.; Ulasov, I.V.; Borovjagin, A.; Sonabend, A.M.; Khramtsov, A.; Han, Y.; Dent, P.; Fisher, P.B.; Curiel, D.T.; Lesniak, M.S. Enhanced transduction of malignant glioma with a double targeted Ad5/3-RGD fiber-modified adenovirus. Mol. Cancer Ther. 2006, 5, 2408–2416.
- Tyler, M.A.; Ulasov, I.V.; Borovjagin, A.; Sonabend, A.M.; Khramtsov, A.; Han, Y.; Dent, P.; Fisher, P.B.; Curiel, D.T.; Lesniak, M.S. Enhanced transduction of malignant glioma with a double targeted Ad5/3-RGD fiber-modified adenovirus. Mol. Cancer Ther. 2006, 5, 2408–2416.Shayakhmetov, D.M.; Eberly, A.M.; Li, Z.; Lieber, A. Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J. Virol. 2005, 79, 1053–1061.
- Shayakhmetov, D.M.; Eberly, A.M.; Li, Zo.; Lieber, A. Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J. Virol. 2005, 79, 1053–1061.Martinez-Velez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Pérez-Larraya, J.G.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspide, V. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10, 2235.
- Martinez-Velez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Pérez-Larraya, J.G.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspide, V. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10, 2235.Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427.
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427.Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854.
- Meckes, D.G.; Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol.2011, 85, 12844–12854.Yao, X.L.; Yoshioka, Y.; Ruan, G.; Chen, Y.; Mizuguchi, H.; Mukai, Y.; Okada, N.; Gao, J.; Nakagawa, S. Optimization and internalization mechanisms of PEGylated adenovirus vector with targeting peptide for cancer gene therapy. Biomacromolecules 2012, 13, 2402–2409.
- Yao, X.L.; Yoshioka, Y.; Ruan, G.; Chen, Y.; Mizuguchi, H.; Mukai, Y.; Okada, N.; Gao, Ji.; Nakagawa, S. Optimization and internalization mechanisms of PEGylated adenovirus vector with targeting peptide for cancer gene therapy. Biomacromolecules 2012, 13, 2402–2409.Nguyen, T.V.; Anguiano-Zarate, S.S.; Matchett, W.E.; Barry, M.E.; Barry, M.A. Retargeted and detargeted adenovirus for gene delivery to the muscle. Virology 2018, 514, 118–123.
- Nguyen, T.V.; Anguiano-Zarate, S.S.; Matchett, W.E.; Barry, M.E.; Barry, M.A. Retargeted and detargeted adenovirus for gene delivery to the muscle. Virology 2018, 514, 118–123.Ghosh, D.; Barry, M.A. Selection of muscle-binding peptides from context-specific peptide-presenting phage libraries for adenoviral vector targeting. J. Virol. 2005, 79, 13667–13672.
- Ghosh, D.; Barry, M.A. Selection of muscle-binding peptides from context-specific peptide-presenting phage libraries for adenoviral vector targeting. J. Virol.2005, 79, 13667–13672.Shashkova, E.V.; May, S.M.; Doronin, K.; Barry, M.A. Expanded anticancer therapeutic window of hexon-modified oncolytic adenovirus. Mol. Ther. 2009, 17, 2121–2130.
- Shashkova, E.V.; May, S.M.; Doronin, K.; Barry. M.A. Expanded anticancer therapeutic window of hexon-modified oncolytic adenovirus. Mol. Ther. 2009, 17, 2121–2130.Shashkova, E.V.; Doronin, K.; Senac, J.S.; Barry, M.A. Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res. 2008, 68, 5896–5904.
- Shashkova, E.V.; Doronin, K.; Senac, J.S.; Barry, M.A. Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res. 2008, 68, 5896–5904.de Vrij, J.; Dautzenberg, I.J.C.; van den Hengel, S.K.; Magnusson, M.K.; Uil, T.G.; Cramer, S.J.; Vellinga, J.; Verissimo, C.S.; Lindholm, L.; Koppers-Lalic, D. A cathepsin-cleavage site between the adenovirus capsid protein IX and a tumor-targeting ligand improves targeted transduction. Gene Ther. 2012, 19, 899–906.
- de Vrij, J.; Dautzenberg, I.J.C.; van den Hengel, S.K.; Magnusson, M.K.; Uil, T.G.; Cramer, S.J.; Vellinga, J.; Verissimo, C.S.; Lindholm, L.; Koppers-Lalic, D. A cathepsin-cleavage site between the adenovirus capsid protein IX and a tumor-targeting ligand improves targeted transduction. Gene Ther. 2012, 19, 899–906.de Vrij, J.; Uil, T.G.; van den Hengel, S.K.; Cramer, S.J.; Koppers-Lalic, D.; Verweij, M.C.; Wiertz, E.J.H.J.; Vellinga, J.; Willemsen, R.A.; Hoeben, R.C. Adenovirus targeting to HLA-A1/MAGE-A1-positive tumor cells by fusing a single-chain T-cell receptor with minor capsid protein IX. Gene Ther. 2008, 15, 978–989.
- de Vrij, J.; Uil, T.G.; van den Hengel, S.K.; Cramer, S.J.; Koppers-Lalic, D.; Verweij, M.C.; Wiertz, E.J.H.J.; Vellinga, J.; Willemsen, R.A.; Hoeben, R.C. Adenovirus targeting to HLA-A1/MAGE-A1-positive tumor cells by fusing a single-chain T-cell receptor with minor capsid protein IX. Gene Ther. 2008, 15, 978–989.Kirby, I.; Davison, E.; Beavil, A.J.; Soh, C.P.; Wickham, T.J.; Roelvink, P.W.; Kovesdi, I.; Sutton, B.J.; Santis, G. Identification of contact residues and definition of the CAR-binding site of adenovirus type 5 fiber protein. J. Virol. 2000, 74, 2804–2813.
- Kirby, I.; Davison, E.; Beavil, A.J.; Soh, C.P.; Wickham, T.J.; Roelvink, P.W.; Kovesdi, I.; Sutton, B.J.; Santis, G. Identification of contact residues and definition of the CAR-binding site of adenovirus type 5 fiber protein. J. Virol. 2000, 74, 2804–2813.Kurachi, S.; Tashiro, K.; Sakurai, F.; Sakurai, H.; Kawabata, K.; Yayama, K.; Okamoto, H.; Nakagawa, S.; Mizuguchi, H. Fiber-modified adenovirus vectors containing the TAT peptide derived from HIV-1 in the fiber knob have efficient gene transfer activity. Gene Ther. 2007, 14, 1160–1165.
- Kurachi, S.; Tashiro, K.; Sakurai, F.; Sakurai, H.; Kawabata, K.; Yayama, K.; Okamoto, H.; Nakagawa, S.; Mizuguchi, H. Fiber-modified adenovirus vectors containing the TAT peptide derived from HIV-1 in the fiber knob have efficient gene transfer activity. Gene Ther. 2007, 14, 1160–1165.Schmidt, A.; Eipel, C.; Fürst, K.; Sommer, N.; Pahnke, J.; Pützer, B.M. Evaluation of systemic targeting of RET oncogene-based MTC with tumor-selective peptide-tagged Ad vectors in clinical mouse models. Gene Ther. 2011, 18, 418–423.
- Schmidt, A.; Eipel, C.; Fürst, K.; Sommer, N.; Pahnke, J.; Pützer, B.M. Evaluation of systemic targeting of RET oncogene-based MTC with tumor-selective peptide-tagged Ad vectors in clinical mouse models. Gene Ther. 2011, 18, 418–423.Reetz, J.; Herchenroder, O.; Putzer, B.M. Peptide-based technologies to alter adenoviral vector tropism: Ways and means for systemic treatment of cancer. Viruses 2014, 6, 1540–1563.
- Reetz, J.; Herchenroder, O.; Putzer, B.M. Peptide-based technologies to alter adenoviral vector tropism: Ways and means for systemic treatment of cancer. Viruses 2014, 6, 1540–1563.Belousova, N.; Korokhov, N.; Krendelshchikova, V.; Simonenko, V.; Mikheeva, G.; Triozzi, P.L.; Aldrich, W.A.; Banerjee, P.T.; Gillies, S.D.; Curiel, D.T. Genetically targeted adenovirus vector directed to CD40-expressing cells. J. Virol. 2003, 77, 11367–11377.
- Belousova, N.; Korokhov, N.; Krendelshchikova, V.; Simonenko, V.; Mikheeva, G.; Triozzi, P.L.; Aldrich, W.A.; Banerjee, P.T.; Gillies, S.D.; Curiel, D.T. Genetically targeted adenovirus vector directed to CD40-expressing cells. J. Virol. 2003, 77, 11367–11377.Nicklin, S.A.; White, S.J.; Watkins, S.J.; Hawkins, R.E.; Baker, A.H. Selective targeting of gene transfer to vascular endothelial cells by use of peptides isolated by phage display. Circulation 2000, 102, 231–237.
- Nicklin, S.A.; White, S.J.; Watkins, S.J.; Hawkins, R.E.; Baker, A.H. Selective targeting of gene transfer to vascular endothelial cells by use of peptides isolated by phage display. Circulation 2000, 102, 231–237.Xia, H.; Anderson, B.; Mao, Q.; Davidson, B.L. Recombinant human adenovirus: Targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J. Virol. 2000, 74, 11359–11366.
- Xia, H.; Anderson, B.; Mao, Q.; Davidson, B.L. Recombinant human adenovirus: Targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J. Virol. 2000, 74, 11359–11366.Schmidt, A.; Haas, S.J.-; Hildebrandt, S.; Scheibe, J.; Eckhoff, B.; Racek, T.; Kempermann, G.; Wree, A.; Pützer, B.M. Selective targeting of adenoviral vectors to neural precursor cells in the hippocampus of adult mice: New prospects for in situ gene therapy. Stem Cells 2007, 25, 2910–2918.
- Schmidt, A.; Haas, S.J.-; Hildebrandt, S.; Scheibe, J.; Eckhoff, B.; Racek, T.; Kempermann, G.; Wree, A.; Pützer, B.M. Selective targeting of adenoviral vectors to neural precursor cells in the hippocampus of adult mice: New prospects for in situ gene therapy. Stem Cells 2007, 25, 2910–2918.Dmitriev, I.; Krasnykh, V.; Miller, C.R.; Wang, M.; Kashentseva, E.; Mikheeva, G.; Belousova, N.; Curiel, D.T. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J. Virol. 1998, 72, 9706–9713.
- Dmitriev, I.; Krasnykh, V.; Miller, C.R.; Wang, M.; Kashentseva, E.; Mikheeva, G.; Belousova, N.; Curiel, D.T. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J. Virol. 1998, 72, 9706–9713.Nicklin, S.A.; Dishart, K.L.; Buening, H.; Reynolds, P.N.; Hallek, M.; Nemerow, G.R.; von Seggern, D.J.; Baker, A.H. Transductional and transcriptional targeting of cancer cells using genetically engineered viral vectors. Cancer Lett. 2003, 201, 165–173.
- Nicklin, S.A.; Dishart, K.L.; Buening, H.; Reynolds, P.N.; Hallek, M.; Nemerow, G.R.; von Seggern, D.J.; Baker, A.H. Transductional and transcriptional targeting of cancer cells using genetically engineered viral vectors. Cancer Lett. 2003, 201, 165–173.Work, L.M.; Nicklin, S.A.; Brain, N.J.R.; Dishart, K.L.; von Seggern, D.J.; Hallek, M.; Büning, H.; Baker, A.H. Development of efficient viral vectors selective for vascular smooth muscle cells. Mol. Ther. 2004, 9, 198–208.
- Work, L.M.; Nicklin, S.A.; Brain, N.J.R.; Dishart, K.L.; von Seggern, D.J.; Hallek, M.; Büning, H.; Baker, A.H. Development of efficient viral vectors selective for vascular smooth muscle cells. Mol. Ther. 2004, 9, 198–208.Yoon, A.R.; Hong, J.; Kim, S.W.; Yun, C.-O. Redirecting adenovirus tropism by genetic, chemical, and mechanical modification of the adenovirus surface for cancer gene therapy. Expert Opin. Drug Deliv. 2016, 13, 843–858.
- Yoon, A.R.; Hong, J.; Kim, S.W.; Yun, C.-O. Redirecting adenovirus tropism by genetic, chemical, and mechanical modification of the adenovirus surface for cancer gene therapy. Expert Opin. Drug Deliv. 2016, 13, 843–858.Nishimoto, T.; Yoshida, K.; Miura, Y.; Kobayashi, A.; Hara, H.; Ohnami, S.; Kurisu, K.; Yoshida, T.; Aoki, K. Oncolytic virus therapy for pancreatic cancer using the adenovirus library displaying random peptides on the fiber knob. Gene Ther. 2009, 16, 669–680.
- Nishimoto, T.; Yoshida, K.; Miura, Y.; Kobayashi, A.; Hara, H.; Ohnami, S.; Kurisu, K.; Yoshida, T.; Aoki, K. Oncolytic virus therapy for pancreatic cancer using the adenovirus library displaying random peptides on the fiber knob. Gene Ther. 2009, 16, 669–680.Miura, Y.; Yoshida, K.; Nishimoto, T.; Hatanaka, K.; Ohnami, S.; Asaka, M.; Douglas, J.T.; Curiel, D.T.; Yoshida, T.; Aoki, K. Direct selection of targeted adenovirus vectors by random peptide display on the fiber knob. Gene Ther. 2007, 14, 1448–1460.
- Miura, Y.; Yoshida, K.; Nishimoto, T.; Hatanaka, K.; Ohnami, S.; Asaka, M.; Douglas, J.T.; Curiel, D.T.; Yoshida, T.; Aoki, K. Direct selection of targeted adenovirus vectors by random peptide display on the fiber knob. Gene Ther. 2007, 14, 1448–1460.Belousova, N.; Krendelchtchikova, V.; Curiel, D.T.; Krasnykh, V. Modulation of adenovirus vector tropism via incorporation of polypeptide ligands into the fiber protein. J. Virol. 2002, 76, 8621–8631.
- Belousova, N.; Krendelchtchikova, V.; Curiel, D.T.; Krasnykh, V. Modulation of adenovirus vector tropism via incorporation of polypeptide ligands into the fiber protein. J. Virol. 2002, 76, 8621–8631.Wickham, T.J.; Tzeng, E.; Shears, L.L., 2nd; Roelvink, P.W.; Li, Y.; Lee, G.M.; Brough, D.E.; Lizonova, A.; Kovesdi, I. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. J. Virol. 1997, 71, 8221–8229.
- Wickham, T.J.; Tzeng, E.; Shears, L.L., 2nd; Roelvink, P.W.; Li, Y.; Lee, G.M.; Brough, D.E.; Lizonova, A.; Kovesdi, I. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. J. Virol. 1997, 71, 8221–8229.Sato-Dahlman, M.; Miura, Y.; Huang, J.L.; Hajeri, P.; Jacobsen, K.; Davydova, J.; Yamamoto, M. CD133-targeted oncolytic adenovirus demonstrates anti-tumor effect in colorectal cancer. Oncotarget 2017, 8, 76044–76056.
- Sato-Dahlman, M.; Miura, Y.; Huang, J.L.; Hajeri, P.; Jacobsen, K.; Davydova, J.; Yamamoto, M. CD133-targeted oncolytic adenovirus demonstrates anti-tumor effect in colorectal cancer. Oncotarget 2017, 8, 76044–76056.Beatty, M.S.; Curiel, D.T. Chapter two—Adenovirus strategies for tissue-specific targeting. Adv. Cancer Res. 2012, 115, 39–67.
- Beatty, M.S.; Curiel, D.T. Chapter two—Adenovirus strategies for tissue-specific targeting. Adv. Cancer Res. 2012, 115, 39–67.Parrott, M.B.; Adams, K.E.; Mercier, G.T.; Mok, H.; Campos, S.K.; Barry, M.A. Metabolically biotinylated adenovirus for cell targeting, ligand screening, and vector purification. Mol. Ther. 2003, 8, 688–700.
- Parrott, M.B.; Adams, K.E.; Mercier, G.T.; Mok, H.; Campos, S.K.; Barry, M.A. Metabolically biotinylated adenovirus for cell targeting, ligand screening, and vector purification. Mol. Ther. 2003, 8, 688–700.Pereboeva, L.; Komarova, S.; Roth, J.; Ponnazhagan, S.; Curiel, D.T. Targeting EGFR with metabolically biotinylated fiber-mosaic adenovirus. Gene Ther. 2007, 14, 627–637.
- Pereboeva, L.; Komarova, S.; Roth, J.; Ponnazhagan, S.; Curiel, D.T. Targeting EGFR with metabolically biotinylated fiber-mosaic adenovirus. Gene Ther. 2007, 14, 627–637.Campos, S.K.; Parrott, M.B.; Barry, M.A. Avidin-based targeting and purification of a protein IX-modified, metabolically biotinylated adenoviral vector. Mol. Ther. 2004, 9, 942–954.
- Campos, S.K.; Parrott, M.B.; Barry, M.A. Avidin-based targeting and purification of a protein IX-modified, metabolically biotinylated adenoviral vector. Mol. Ther. 2004, 9, 942–954.Chen, Z.; Mok, H.; Pflugfelder, S.C.; Li, D.Q.; Barry, M.A. Improved transduction of human corneal epithelial progenitor cells with cell-targeting adenoviral vectors. Exp. Eye Res. 2006, 83, 798–806.
- Chen, Z.; Mok, H.; Pflugfelder, S.C.; Li, D.Q.; Barry, M.A. Improved transduction of human corneal epithelial progenitor cells with cell-targeting adenoviral vectors. Experimental eye research 2006, 83, 798–806, doi:10.1016/j.exer.2006.03.023.Korokhov, N.; Mikheeva, G.; Krendelshchikov, A.; Belousova, N.; Simonenko, V.; Krendelshchikova, V.; Pereboev, A.; Kotov, A.; Kotova, O.; Triozzi, P.L.; et al. Targeting of adenovirus via genetic modification of the viral capsid combined with a protein bridge. J. Virol. 2003, 77, 12931–12940.
- Korokhov, N.; Mikheeva, G.; Krendelshchikov, A.; Belousova, N.; Simonenko, V.; Krendelshchikova, V.; Pereboev, A.; Kotov, A.; Kotova, O.; Triozzi, P.L., et al. Targeting of adenovirus via genetic modification of the viral capsid combined with a protein bridge. J Virol 2003, 77, 12931–12940, doi:10.1128/jvi.77.24.12931-12940.2003.Volpers, C.; Thirion, C.; Biermann, V.; Hussmann, S.; Kewes, H.; Dunant, P.; von der Mark, H.; Herrmann, A.; Kochanek, S.; Lochmuller, H. Antibody-mediated targeting of an adenovirus vector modified to contain a synthetic immunoglobulin g-binding domain in the capsid. J. Virol. 2003, 77, 2093–2104.
- Volpers, C.; Thirion, C.; Biermann, V.; Hussmann, S.; Kewes, H.; Dunant, P.; von der Mark, H.; Herrmann, A.; Kochanek, S.; Lochmuller, H. Antibody-mediated targeting of an adenovirus vector modified to contain a synthetic immunoglobulin g-binding domain in the capsid. J Virol 2003, 77, 2093–2104, doi:10.1128/jvi.77.3.2093-2104.2003.Wickham, T.J.; Lee, G.M.; Titus, J.A.; Sconocchia, G.; Bakacs, T.; Kovesdi, I.; Segal, D.M. Targeted adenovirus-mediated gene delivery to T cells via CD3. J. Virol. 1997, 71, 7663–7669.
- Wickham, T.J.; Lee, G.M.; Titus, J.A.; Sconocchia, G.; Bakacs, T.; Kovesdi, I.; Segal, D.M. Targeted adenovirus-mediated gene delivery to T cells via CD3. J Virol 1997, 71, 7663–7669.Borovjagin, A.V.; Krendelchtchikov, A.; Ramesh, N.; Yu, D.C.; Douglas, J.T.; Curiel, D.T. Complex mosaicism is a novel approach to infectivity enhancement of adenovirus type 5-based vectors. Cancer Gene Ther. 2005, 12, 475–486.
- Borovjagin, A.V.; Krendelchtchikov, A.; Ramesh, N.; Yu, D.C.; Douglas, J.T.; Curiel, D.T. Complex mosaicism is a novel approach to infectivity enhancement of adenovirus type 5-based vectors. Cancer Gene Ther. 2005, 12, 475–486, doi:10.1038/sj.cgt.7700806.Sakurai, F. Development and evaluation of a novel gene delivery vehicle composed of adenovirus serotype 35. Biol. Pharm. Bull. 2008, 31, 1819–1825.
- Sakurai, F. Development and evaluation of a novel gene delivery vehicle composed of adenovirus serotype 35. Biol. Pharm. Bull. 2008, 31, 1819–1825, doi:10.1248/bpb.31.1819.Preuss, M.A.; Glasgow, J.N.; Everts, M.; Stoff-Khalili, M.A.; Wu, H.; Curiel, D.T. Enhanced Gene Delivery to Human Primary Endothelial Cells Using Tropism-Modified Adenovirus Vectors. Open Gene Ther. J. 2008, 1, 7–11.
- Preuss, M.A.; Glasgow, J.N.; Everts, M.; Stoff-Khalili, M.A.; Wu, H.; Curiel, D.T. Enhanced Gene Delivery to Human Primary Endothelial Cells Using Tropism-Modified Adenovirus Vectors. Open Gene Ther. J. 2008, 1, 7–11, doi:10.2174/1875037000801010007.Yang, M.; Yang, C.S.; Guo, W.; Tang, J.; Huang, Q.; Feng, S.; Jiang, A.; Xu, X.; Jiang, G.; Liu, Y.Q. A novel fiber chimeric conditionally replicative adenovirus-Ad5/F35 for tumor therapy. Cancer Biol. Ther. 2017, 18, 833–840.
- Yang, M.; Yang, C.S.; Guo, W.; Tang, J.; Huang, Q.; Feng, S.; Jiang, A.; Xu, X.; Jiang, G.; Liu, Y.Q. A novel fiber chimeric conditionally replicative adenovirus-Ad5/F35 for tumor therapy. Cancer Biol. Ther. 2017, 18, 833–840, doi:10.1080/15384047.2017.1395115.Kanerva, A.; Mikheeva, G.V.; Krasnykh, V.; Coolidge, C.J.; Lam, J.T.; Mahasreshti, P.J.; Barker, S.D.; Straughn, M.; Barnes, M.N.; Alvarez, R.D.; et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin Cancer Res 2002, 8, 275–280.
- Kanerva, A.; Mikheeva, G.V.; Krasnykh, V.; Coolidge, C.J.; Lam, J.T.; Mahasreshti, P.J.; Barker, S.D.; Straughn, M.; Barnes, M.N.; Alvarez, R.D., et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin Cancer Res 2002, 8, 275–280.Guse, K.; Ranki, T.; Ala-Opas, M.; Bono, P.; Sarkioja, M.; Rajecki, M.; Kanerva, A.; Hakkarainen, T.; Hemminki, A. Treatment of metastatic renal cancer with capsid-modified oncolytic adenoviruses. Mol. Cancer Ther. 2007, 6, 2728–2736.
- Guse, K.; Ranki, T.; Ala-Opas, M.; Bono, P.; Sarkioja, M.; Rajecki, M.; Kanerva, A.; Hakkarainen, T.; Hemminki, A. Treatment of metastatic renal cancer with capsid-modified oncolytic adenoviruses. Mol. Cancer The. 2007, 6, 2728–2736, doi:10.1158/1535-7163.MCT-07-0176.Wei, X.; Liu, L.; Wang, G.; Li, W.; Xu, K.; Qi, H.; Liu, H.; Shen, J.; Li, Z.; Shao, J. Potent antitumor activity of the Ad5/11 chimeric oncolytic adenovirus combined with interleukin-24 for acute myeloid leukemia via induction of apoptosis. Oncol. Rep. 2015, 33, 111–118.
- Wei, X.; Liu, L.; Wang, G.; Li, W.; Xu, K.; Qi, H.; Liu, H.; Shen, J.; Li, Z.; Shao, J. Potent antitumor activity of the Ad5/11 chimeric oncolytic adenovirus combined with interleukin-24 for acute myeloid leukemia via induction of apoptosis. Oncol. Rep. 2015, 33, 111–118, doi:10.3892/or.2014.3563.Machiels, J.P.; Salazar, R.; Rottey, S.; Duran, I.; Dirix, L.; Geboes, K.; Wilkinson-Blanc, C.; Pover, G.; Alvis, S.; Champion, B.; et al. A phase 1 dose escalation study of the oncolytic adenovirus enadenotucirev, administered intravenously to patients with epithelial solid tumors (EVOLVE). J. Immunother. Cancer 2019, 7, 20.
- Machiels, J.P.; Salazar, R.; Rottey, S.; Duran, I.; Dirix, L.; Geboes, K.; Wilkinson-Blanc, C.; Pover, G.; Alvis, S.; Champion, B., et al. A phase 1 dose escalation study of the oncolytic adenovirus enadenotucirev, administered intravenously to patients with epithelial solid tumors (EVOLVE). J. Immunother. Cancer 2019, 7, 20, doi:10.1186/s40425-019-0510-7.Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884.
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K., et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther 2010, 18, 1874–1884, doi:10.1038/mt.2010.161.Kim, K.H.; Dmitriev, I.P.; Saddekni, S.; Kashentseva, E.A.; Harris, R.D.; Aurigemma, R.; Bae, S.; Singh, K.P.; Siegal, G.P.; Curiel, D.T.; et al. A phase I clinical trial of Ad5/3-Delta24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol. Oncol. 2013, 130, 518–524.
- Kim, K.H.; Dmitriev, I.P.; Saddekni, S.; Kashentseva, E.A.; Harris, R.D.; Aurigemma, R.; Bae, S.; Singh, K.P.; Siegal, G.P.; Curiel, D.T., et al. A phase I clinical trial of Ad5/3-Delta24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol. Oncol. 2013, 130, 518–524., doi:10.1016/j.ygyno.2013.06.003.Seki, T.; Dmitriev, I.; Suzuki, K.; Kashentseva, E.; Takayama, K.; Rots, M.; Uil, T.; Wu, H.; Wang, M.; Curiel, D.T. Fiber shaft extension in combination with HI loop ligands augments infectivity for CAR-negative tumor targets but does not enhance hepatotropism in vivo. Gene Ther. 2002, 9, 1101–1108.
- Seki, T.; Dmitriev, I.; Suzuki, K.; Kashentseva, E.; Takayama, K.; Rots, M.; Uil, T.; Wu, H.; Wang, M.; Curiel, D.T. Fiber shaft extension in combination with HI loop ligands augments infectivity for CAR-negative tumor targets but does not enhance hepatotropism in vivo. Gene Ther. 2002, 9, 1101–1108., doi:10.1038/sj.gt.3301815.Janzen, W. High Throughput Screening: Methods and Protocols; Humana Press: New York, NY, USA, 2016; Volume 1439.
- Wu, P.; Kudrolli, T.A.; Chowdhury, W.H.; Liu, M.M.; Rodriguez, R.; Lupold, S.E. Adenovirus targeting to prostate-specific membrane antigen through virus-displayed, semirandom peptide library screening. Cancer Res. 2010, 70, 9549–9553.
- Janzen, W. High Throughput Screening: Methods and Protocols; Humana Press: New York, NY, USA, 2016; Volume 1439.Hust, M.; Lim, T.S. Phage Display: Methods and Protocols; Humana Press: New York, NY, USA, 2018.
- Hust, M.; Lim, T.S. Phage display: Methods and Protocols; Humana Press: New York, NY, USA, 2018.Lupold, S.E.; Kudrolli, T.A.; Chowdhury, W.H.; Wu, P.; Rodriguez, R. A novel method for generating and screening peptides and libraries displayed on adenovirus fiber. Nucleic Acids Res. 2007, 35, e138.
- Lupold, S.E.; Kudrolli, T.A.; Chowdhury, W.H.; Wu, P.; Rodriguez, R. A novel method for generating and screening peptides and libraries displayed on adenovirus fiber. Nucleic Acids Res. 2007, 35, e138.Weber, M.; Bujak, E.; Putelli, A.; Villa, A.; Matasci, M.; Gualandi, L.; Hemmerle, T.; Wulhfard, S.; Neri, D. A highly functional synthetic phage display library containing over 40 billion human antibody clones. PLoS ONE 2014, 9, e100000.
- Weber, M.; Bujak, E.; Putelli, A.; Villa, A.; Matasci, M.; Gualandi, L.; Hemmerle, T.; Wulhfard, S.; Neri, D. A highly functional synthetic phage display library containing over 40 billion human antibody clones. PLoS ONE 2014, 9, e100000.Matochko, W.L.; Chu, K.; Jin, B.; Lee, S.W.; Whitesides, G.M.; Derda, R. Deep sequencing analysis of phage libraries using Illumina platform. Methods 2012, 58, 47–55.
- Matochko, W.L.; Chu, K.; Jin, B.; Lee, S.W.; Whitesides, G.M.; Derda, R. Deep sequencing analysis of phage libraries using Illumina platform. Methods 2012, 58, 47–55.Hajeri, P.B.; Yamamoto, M. A Novel Method to Generate HighSequence Diversity Libraries of Plasmids and Recombinant Adenovirus Based Oncolytic Viruses for Targeted Therapy 2020 ASGCT Annual Meeting Abstracts. Mol. Ther. 2020, 28, 1–592.
- Hajeri, P.B.; Yamamoto, M. A Novel Method to Generate HighSequence Diversity Libraries of Plasmids and Recombinant Adenovirus Based Oncolytic Viruses for Targeted Therapy 2020 ASGCT Annual Meeting Abstracts. Mol. Ther. 2020, 28, 1–592.Haisma, H.J.; Grill, J.; Curiel, D.T.; Hoogeland, S.; van Beusechem, V.W.; Pinedo, H.M.; Gerritsen, W.R. Targeting of adenoviral vectors through a bispecific single-chain antibody. Cancer Gene Ther. 2000, 7, 901–904.
- Haisma, H.J.; Grill, J.; Curiel, D.T.; Hoogeland, S.; van Beusechem, V.W.; Pinedo, H.M.; Gerritsen, W.R. Targeting of adenoviral vectors through a bispecific single-chain antibody. Cancer Gene Ther. 2000, 7, 901–904.Haisma, H.J.; Kamps, G.K.; Bouma, A.; Geel, T.M.; Rots, M.G.; Kariath, A.; Bellu, A.R. Selective targeting of adenovirus to alphavbeta3 integrins, VEGFR2 and Tie2 endothelial receptors by angio-adenobodies. Int. J. Pharm. 2010, 391, 155–161.
- Haisma, H.J.; Kamps, G.K.; Bouma, A.; Geel, T.M.; Rots, M.G.; Kariath, A.; Bellu, A.R. Selective targeting of adenovirus to alphavbeta3 integrins, VEGFR2 and Tie2 endothelial receptors by angio-adenobodies. Int. J. Pharm. 2010, 391, 155–161.Carette, J.E.; Graat, H.C.A.; Schagen, F.H.E.; Mastenbroek, D.C.J.; Rots, M.G.; Haisma, H.J.; Groothuis, G.M.M.; Schaap, G.R.; Bras, J.; Kaspers, G.J.L. A conditionally replicating adenovirus with strict selectivity in killing cells expressing epidermal growth factor receptor. Virology 2007, 361, 56–67.
- Carette, J.E.; Graat, H.C.A.; Schagen, F.H.E.; Mastenbroek, D.C.J.; Rots, M.G.; Haisma, H.J.; Groothuis, G.M.M.; Schaap, G.R.; Bras, J.; Kaspers, G.J.L. A conditionally replicating adenovirus with strict selectivity in killing cells expressing epidermal growth factor receptor. Virology 2007, 361, 56–67.Grill, J.; van Beusechem, V.W.; van der Valk, P.; Dirven, C.M.; Leonhart, A.; Pherai, D.S.; Haisma, H.J.; Pinedo, H.M.; Curiel, D.T.; Gerritsen, W.R. Combined targeting of adenoviruses to integrins and epidermal growth factor receptors increases gene transfer into primary glioma cells and spheroids. Clin. Cancer Res. 2001, 7, 641–650.
- Grill, J.; van Beusechem, V.W.; van der Valk, P.; Dirven, C.M.; Leonhart, A.; Pherai, D.S.; Haisma, H.J.; Pinedo, H.M.; Curiel, D.T.; Gerritsen, W.R. Combined targeting of adenoviruses to integrins and epidermal growth factor receptors increases gene transfer into primary glioma cells and spheroids. Clin. Cancer Res. 2001, 7, 641–650.Nettelbeck, D.M.; Miller, D.W.; Jérôme, V.; Zuzarte, M.; Watkins, S.J.; Hawkins, R.E.; Müller, R.; Kontermann, R.E. Targeting of adenovirus to endothelial cells by a bispecific single-chain diabody directed against the adenovirus fiber knob domain and human endoglin (CD105). Mol. Ther. 2001, 3, 882–891.
- Nettelbeck, D.M.; Miller, D.W.; Jérôme, V.; Zuzarte, M.; Watkins, S.J.; Hawkins, R.E.; Müller, R.; Kontermann, R.E. Targeting of adenovirus to endothelial cells by a bispecific single-chain diabody directed against the adenovirus fiber knob domain and human endoglin (CD105). Mol. Ther. 2001, 3, 882–891.Nettelbeck, D.M.; Rivera, A.A.; Kupsch, J.; Dieckmann, D.; Douglas, J.T.; Kontermann, R.E.; Alemany, R.; Curiel, D.T. Retargeting of adenoviral infection to melanoma: Combining genetic ablation of native tropism with a recombinant bispecific single-chain diabody (scDb) adapter that binds to fiber knob and HMWMAA. Int. J. Cancer 2004, 108, 136–145.
- Nettelbeck, D.M.; Rivera, A.A.; Kupsch, J.; Dieckmann, D.; Douglas, J.T.; Kontermann, R.E.; Alemany, R.; Curiel, D.T. Retargeting of adenoviral infection to melanoma: Combining genetic ablation of native tropism with a recombinant bispecific single-chain diabody (scDb) adapter that binds to fiber knob and HMWMAA. Int. J. Cancer 2004, 108, 136–145.Douglas, J.T.; Rogers, B.E.; Rosenfeld, M.E.; Michael, S.I.; Feng, M.; Curiel, D.T. Targeted gene delivery by tropism-modified adenoviral vectors. Nat. Biotechnol. 1996, 14, 1574–1578.
- Douglas, J.T.; Rogers, B.E.; Rosenfeld, M.E.; Michael, S.I.; Feng, M.; Curiel, D.T. Targeted gene delivery by tropism-modified adenoviral vectors. Nat. Biotechnol. 1996, 14, 1574–1578.Li, E.; Brown, S.L.; von Seggern, D.J.; Brown, G.B.; Nemerow, G.R. Signaling antibodies complexed with adenovirus circumvent CAR and integrin interactions and improve gene delivery. Gene Ther. 2000, 7, 1593–1599.
- Li, E.; Brown, S.L.; von Seggern, D.J.; Brown, G.B.; Nemerow, G.R. Signaling antibodies complexed with adenovirus circumvent CAR and integrin interactions and improve gene delivery. Gene Ther. 2000, 7, 1593–1599.Reetz, J.; Genz, B.; Meier, C.; Kowtharapu, B.S.; Timm, F.; Vollmar, B.; Herchenröder, O.; Abshagen, K.; Pützer, B.M. Development of Adenoviral Delivery Systems to Target Hepatic Stellate Cells In Vivo. PLoS ONE 2013, 8, e67091.
- Cherry, T.; Longo, S.L.; Tovar-Spinoza, Z.; Post, D.E. Second-generation HIF-activated oncolytic adenoviruses with improved replication, oncolytic, and antitumor efficacy. Gene Ther. 2010, 17, 1430–1441.Bhatia, S.; O’Bryan, S.M.; Rivera, A.A.; Curiel, D.T.; Mathis, J.M. CXCL12 retargeting of an adenovirus vector to cancer cells using a bispecific adapter. Oncolytic Virother. 2016, 5, 99–113.
- Post, D.E.; Sandberg, E.M.; Kyle, M.M.; Devi, N.S.; Brat, D.J.; Xu, Z.; Tighiouart, M.; van Meir, E.G. Targeted cancer gene therapy using a hypoxia inducible factor dependent oncolytic adenovirus armed with interleukin-4. Cancer Res. 2007, 67, 6872–6881.Liang, Q.; Dmitriev, I.; Kashentseva, E.; Curiel, D.T.; Herschman, H.R. Noninvasive of adenovirus tumor retargeting in living subjects by a soluble adenovirus receptor-epidermal growth factor (sCAR-EGF) fusion protein. Mol. Imaging Biol. 2004, 6, 385–394.
- Tazawa, H.; Hasei, J.; Yano, S.; Kagawa, S.; Ozaki, T.; Fujiwara, T. Bone and Soft-Tissue Sarcoma: A New Target for Telomerase-Specific Oncolytic Virotherapy. Cancers 2020, 12, 478.Dmitriev, I.; Kashentseva, E.; Rogers, B.E.; Krasnykh, V.; Curiel, D.T. Ectodomain of coxsackievirus and adenovirus receptor genetically fused to epidermal growth factor mediates adenovirus targeting to epidermal growth factor receptor-positive cells. J. Virol. 2000, 74, 6875–6884.
- Cui, C.X.; Li, Y.-Q.; Sun, Y.-J.; Zhu, Y.-L.; Fang, J.-B.; Bai, B.; Li, W.-J.; Li, S.-Z.; Ma, Y.-Z.; Li, X. Antitumor effect of a dual cancer-specific oncolytic adenovirus on prostate cancer PC-3 cells. Urol. Oncol. 2019, 37, 352 e1–352 e18.Wesseling, J.G.; Bosma, P.J.; Krasnykh, V.; Kashentseva, E.A.; Blackwell, J.L.; Reynolds, P.N.; Li, H.; Parameshwar, M.; Vickers, S.M.; Jaffee, E.M. Improved gene transfer efficiency to primary and established human pancreatic carcinoma target cells via epidermal growth factor receptor and integrin-targeted adenoviral vectors. Gene Ther. 2001, 8, 969–976.
- Lin, J.; Zeng, D.; He, H.; Tan, G.; Lan, Y.; Jiang, F.; Sheng, S. Gene therapy for human ovarian cancer cells using efficient expression of Fas gene combined with gammadeltaT cells. Mol. Med. Rep. 2017, 16, 3791–3798.Pereboev, A.V.; Asiedu, C.K.; Kawakami, Y.; Dong, S.S.; Blackwell, J.L.; Kashentseva, E.A.; Triozzi, P.L.; Aldrich, W.A.; Curiel, D.T.; Thomas, J.M. Coxsackievirus-adenovirus receptor genetically fused to anti-human CD40 scFv enhances adenoviral transduction of dendritic cells. Gene Ther. 2002, 9, 1189–1193.
- Kim, Y.H.; Kim, K.T.; Lee, S.A.; Hong, S.E.; Moon, J.Y.; Yoon, E.K.; Kim, S.; Kim, E.O.; Kang, S.H.; Kim, S.K. Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics 2016, 6, 357–368.Kim, J.; Smith, T.; Idamakanti, N.; Mulgrew, K.; Kaloss, M.; Kylefjord, H.; Ryan, P.C.; Kaleko, M.; Stevenson, S.C. Targeting adenoviral vectors by using the extracellular domain of the coxsackie-adenovirus receptor: Improved potency via trimerization. J. Virol. 2002, 76, 1892–1903.
- Wu, X.; Chen, J.; Cao, Y.; Xie, B.; Li, H.; Zhou, P.; Qiu, Y.; Pang, J. Antitumor effect of COOH-terminal polypeptide of human TERT is associated with the declined expression of hTERT and NF-kappaB p65 in HeLa cells. Oncol. Rep. 2015, 34, 2909–2916.Kashentseva, E.A.; Seki, T.; Curiel, D.T.; Dmitriev, I.P. Adenovirus targeting to c-erbB-2 oncoprotein by single-chain antibody fused to trimeric form of adenovirus receptor ectodomain. Cancer Res. 2002, 62, 609–616.
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 2000, 6, 1134–1139.Kloos, A.; Woller, N.; Gürlevik, E.; Ureche, C.; Niemann, J.; Armbrecht, N.; Martin, N.T.; Geffers, R.; Manns, M.P.; Gerardy-Schahn, R. PolySia-Specific Retargeting of Oncolytic Viruses Triggers Tumor-Specific Immune Responses and Facilitates Therapy of Disseminated Lung Cancer. Cancer Immunol. Res. 2015, 3, 751–763.
- Heise, C.; Sampson-Johannes, A.; Williams, A.; Mccormick, F.; Von Hoff, D.D.; Kirn, D.H. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997, 3, 639–645.Francisco, J.A.; Gilliland, L.K.; Stebbins, M.R.; Norris, N.A.; Ledbetter, J.A.; Siegall, C.B. Activity of a single-chain immunotoxin that selectively kills lymphoma and other B-lineage cells expressing the CD40 antigen. Cancer Res. 1995, 55, 3099–3104.
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12.Chen, C.Y.; May, S.M.; Barry, M.A. Targeting Adenoviruses Factor X-Single-Chain Antibody Fusion Proteins. Hum. Gene Ther. 2010, 21, 739–749.
- Ganesh, S.; Gonzalez-Edick, M.; Gibbons, D.; Ge, Y.; VanRoey, M.; Robinson, M.; Jooss, K. Combination therapy with radiation or cisplatin enhances the potency of Ad5/35 chimeric oncolytic adenovirus in a preclinical model of head and neck cancer. Cancer Gene Ther. 2009, 16, 383–392.Wang, Z.; Yu, B.; Wang, B.; Yan, J.; Feng, X.; Wang, Z.; Wang, L.; Zhang, H.; Wu, H.; Wu, J. A novel capsid-modified oncolytic recombinant adenovirus type 5 for tumor-targeting gene therapy by intravenous route. Oncotarget 2016, 7, 47287–47301.
- Reid, T.R.; Freeman, S.; Post, L.; McCormick, F.; Sze, D.Y. Effects of Onyx-015 among metastatic colorectal cancer patients that have failed prior treatment with 5-FU/leucovorin. Cancer Gene Ther. 2005, 12, 673–681.Garas, M.N.; Tillib, S.V.; Zubkova, O.V.; Rogozhin, V.N.; Ivanova, T.I.; Vasilev, L.A.; Logunov, D.Y.; Shmarov, M.M.; Tutykhina, I.L.; Esmagambetov, I.B. Construction of a pIX-modified Adenovirus Vector Able to Effectively Bind to Nanoantibodies for Targeting. Acta Nat. 2014, 6, 95–105.
- Graat, H.C.; Carette, J.E.; Schagen, F.H.E.; Vassilev, L.T.; Gerritsen, W.R.; Kaspers, G.J.L.; Wuisman, P.I.J.M.; van Beusechem, V.W. Enhanced tumor cell kill by combined treatment with a small-molecule antagonist of mouse double minute 2 and adenoviruses encoding p53. Mol. Cancer Ther. 2007, 6, 1552–1561.Glasgow, J.N.; Mikheeva, G.; Krasnykh, V.; Curiel, D.T. A strategy for adenovirus vector targeting with a secreted single chain antibody. PLoS ONE 2009, 4, e8355.
- Lu, S.; Cullen, B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004, 78, 12868–12876.Jakubczak, J.L.; Rollence, M.L.; Stewart, D.A.; Jafari, J.D.; von Seggern, D.J.; Nemerow, G.R.; Stevenson, S.C.; Hallenbeck, P.L. Adenovirus type 5 viral particles pseudotyped with mutagenized fiber proteins show diminished infectivity of coxsackie B-adenovirus receptor-bearing cells. J. Virol. 2001, 75, 2972–2981.
- Yang, Z.; Li, Y. Dissection of RNAi-based antiviral immunity in plants. Curr. Opin. Virol. 2018, 32, 88–99.Porter, C.E.; Shaw, A.R.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262.
- Schuster, S.; P. Miesen; van Rij, R.P. Antiviral RNAi in Insects and Mammals: Parallels and Differences. Viruses 2019, 11, 448.Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783.
- Zhao, J.H.; Hua, C.L.; Fang, Y.Y.; Guo, H.S. The dual edge of RNA silencing suppressors in the virus-host interactions. Curr. Opin. Virol. 2016, 17, 39–44.Wing, A.; Fajardo, C.A.; Posey, A.D., Jr.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616.
- Machitani, M.; Sakurai, F.; Katayama, K.; Tachibana, M.; Suzuki, T.; Matsui, H.; Yamaguchi, T.; Mizuguchi, H. Improving adenovirus vector-mediated RNAi efficiency by lacking the expression of virus-associated RNAs. Virus Res. 2013, 178, 357–363.Freedman, J.D.; Hagel, J.; Scott, E.M.; Psallidas, I.; Gupta, A.; Spiers, L.; Miller, P.; Kanellakis, N.; Ashfield, R.; Fisher, K.D. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol. Med. 2017, 9, 1067–1087.
- Aparicio, O.; et al. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J. Virol. 2006, 80, 1376–1384.Fajardo, C.A.; Guedan, S.; Rojas, L.A.; Moreno, R.; Arias-Badia, M.; de Sostoa, J.; June, C.H.; Alemany, R. Oncolytic Adenoviral Delivery of an EGFR-Targeting T-cell Engager Improves Antitumor Efficacy. Cancer Res. 2017, 77, 2052–2063.
- Bennasser, Y.; Chable-Bessia, C.; Triboulet, R.; Gibbings, D.; Gwizdek, C.; Dargemont, C.; Kremer, E.J.; Voinnet, O.; Benkirane, M. Competition for XPO5 binding between Dicer mRNA, pre-miRNA and viral RNA regulates human Dicer levels. Nat. Struct. Mol. Biol. 2011, 18, 323–327.Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865.
- Machitani, M.; Yamaguchi, T.; Shimizu, K.; Sakurai, F.; Katayama, K.; Kawabata, K.; Mizuguchi, H. Adenovirus Vector-Derived VA-RNA-Mediated Innate Immune Responses. Pharmaceutics 2011, 3, 338–353.Krasnykh, V.N.; Mikheeva, G.V.; Douglas, J.T.; Curiel, D.T. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J. Virol. 1996, 70, 6839–6846.
- Kitajewski, J.; Schneider, R.J.; Safer, B.; Munemitsu, S.M.; Samuel, C.E.; Thimmappaya, B.; Shenk, T. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 1986, 45, 195–200.Dranoff, G. GM-CSF-secreting melanoma vaccines. Oncogene 2003, 22, 3188–3192.
- Vachon, V.K.; Conn, G.L. Adenovirus VA RNA: An essential pro-viral non-coding RNA. Virus Res. 2016, 212, 39–52.Ramesh, N.; Ge, Y.; Ennist, D.L.; Zhu, M.; Mina, M.; Ganesh, S.; Reddy, P.S.; Yu, D.-C. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor--armed oncolytic adenovirus for the treatment of bladder cancer. Clin. Cancer. Res. 2006, 12, 305–313.
- Carette, J.E.; Overmeer, R.M.; Schagen, F.H.; Alemany, R.; Barski, O.A.; Gerritsen, W.R.; Van Beusechem, V.W. Conditionally replicating adenoviruses expressing short hairpin RNAs silence the expression of a target gene in cancer cells. Cancer Res. 2004, 64, 2663–2667.Flemington, E.K.; Speck, S.H.; Kaelin, W.G., Jr. E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc. Natl. Acad. Sci. USA 1993, 90, 6914–6918.
- Rovira-Rigau, M.; Raimondi, G.; Marín, M.Á.; Gironella, M.; Alemany, R.; Fillat, C. Bioselection Reveals miR-99b and miR-485 as Enhancers of Adenoviral Oncolysis in Pancreatic Cancer. Mol. Ther. 2019, 27, 230–243.Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J. Urol. 2012, 188, 2391–2397.
- Brachtlova, T.; van Beusechem, V.W. Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens. Cells 2018, 7, 288.Gregory, S.M.; Nazir, S.A.; Metcalf, J.P. Implications of the innate immune response to adenovirus and adenoviral vectors. Future Virol. 2011, 6, 357–374.
- Machitani, M.; Sakurai, F.; Wakabayashi, K.; Tachibana, M.; Fujiwara, T.; Mizuguchi, H. Enhanced Oncolytic Activities of the Telomerase-Specific Replication-Competent Adenovirus Expressing Short-Hairpin RNA against Dicer. Mol. Cancer Ther. 2017, 16, 251–259.Tong, A.W.; Stone, M.J. Prospects for Cd40-directed experimental therapy of human cancer. Cancer Gene Ther. 2003, 10, 1–13.
- Reetz, J.; Genz, B.; Meier, C.; Kowtharapu, B.S.; Timm, F.; Vollmar, B.; Herchenröder, O.; Abshagen, K.; Pützer, B.M. Development of Adenoviral Delivery Systems to Target Hepatic Stellate Cells In Vivo. PLoS ONE 2013, 8, e67091.Tong, A.W.; Papayoti, M.H.; Netto, G.; Armstrong, D.T.; Ordonez, G.; Lawson, J.M.; Stone, M.J. Growth-inhibitory effects of CD40 ligand (CD154) and its endogenous expression in human breast cancer. Clin. Cancer Res. 2001, 7, 691–703.
- Bhatia, S.; O'Bryan, S.M.; Rivera, A.A.; Curiel, D.T.; Mathis, J.M. CXCL12 retargeting of an adenovirus vector to cancer cells using a bispecific adapter. Oncolytic Virother. 2016, 5, 99–113.Ghamande, S.; Hylander, B.L.; Oflazoglu, E.; Lele, S.; Fanslow, W.; Repasky, E.A. Recombinant CD40 ligand therapy has significant antitumor effects on CD40-positive ovarian tumor xenografts grown in SCID mice and demonstrates an augmented effect with cisplatin. Cancer Res. 2001, 61, 7556–7562.
- Liang, Q.; Dmitriev, I.; Kashentseva, E.; Curiel, D.T.; Herschman, H.R. Noninvasive of adenovirus tumor retargeting in living subjects by a soluble adenovirus receptor-epidermal growth factor (sCAR-EGF) fusion protein. Mol. Imaging Biol. 2004, 6, 385–394.Nukui, Y.; Picozzi, V.J.; Traverso, L.W. Interferon-based adjuvant chemoradiation therapy improves survival after pancreaticoduodenectomy for pancreatic adenocarcinoma. Am. J. Surg. 2000, 179, 367–371.
- Dmitriev, I.; Kashentseva, E.; Rogers, B.E.; Krasnykh, V.; Curiel, D.T. Ectodomain of coxsackievirus and adenovirus receptor genetically fused to epidermal growth factor mediates adenovirus targeting to epidermal growth factor receptor-positive cells. J. Virol. 2000, 74, 6875–6884.Ohashi, M.; Yoshida, K.; Kushida, M.; Miura, Y.; Ohnami, S.; Ikarashi, Y.; Kitade, Y.; Yoshida, T.; Aoki, K. Adenovirus-mediated interferon alpha gene transfer induces regional direct cytotoxicity and possible systemic immunity against pancreatic cancer. Br. J. Cancer 2005, 93, 441–449.
- Wesseling, J.G.; Bosma, P.J.; Krasnykh, V.; Kashentseva, E.A.; Blackwell, J.L.; Reynolds, P.N.; Li, H.; Parameshwar, M.; Vickers, S.M.; Jaffee, E.M. Improved gene transfer efficiency to primary and established human pancreatic carcinoma target cells via epidermal growth factor receptor and integrin-targeted adenoviral vectors. Gene Ther. 2001, 8, 969–976.Hara, H.; Kobayashi, A.; Yoshida, K.; Ohashi, M.; Ohnami, S.; Uchida, E.; Higashihara, E.; Yoshida, T.; Aoki, K. Local interferon-alpha gene therapy elicits systemic immunity in a syngeneic pancreatic cancer model in hamster. Cancer Sci. 2007, 98, 455–463.
- Pereboev, A.V.; Asiedu, C.K.; Kawakami, Y.; Dong, S.S.; Blackwell, J.L.; Kashentseva, E.A.; Triozzi, P.L.; Aldrich, W.A.; Curiel, D.T.; Thomas, J.M. Coxsackievirus-adenovirus receptor genetically fused to anti-human CD40 scFv enhances adenoviral transduction of dendritic cells. Gene Ther. 2002, 9, 1189–1193.Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Löskog, A. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998.
- Kim, J.; Smith, T.; Idamakanti, N.; Mulgrew, K.; Kaloss, M.; Kylefjord, H.; Ryan, P.C.; Kaleko, M.; Stevenson, S.C. Targeting adenoviral vectors by using the extracellular domain of the coxsackie-adenovirus receptor: Improved potency via trimerization. J. Virol. 2002, 76, 1892–1903.Li, S.; Qi, Z.; Li, H.; Hu, J.; Wang, D.; Wang, X.; Feng, Z. Conditionally replicating oncolytic adenoviral vector expressing arresten and tumor necrosis factor-related apoptosis-inducing ligand experimentally suppresses lung carcinoma progression. Mol. Med. Rep. 2015, 12, 2068–2074.
- Kashentseva, E.A.; Seki, T.; Curiel, D.T.; Dmitriev, I.P. Adenovirus targeting to c-erbB-2 oncoprotein by single-chain antibody fused to trimeric form of adenovirus receptor ectodomain. Cancer Res. 2002, 62, 609–616.Bramante, S.; Kaufmann, J.K.; Veckman, V.; Liikanen, I.; Nettelbeck, D.M.; Hemminki, O.; Vassilev, L.; Cerullo, V.; Oksanen, M.; Heiskanen, R.; et al. Treatment of melanoma with a serotype 5/3 chimeric oncolytic adenovirus coding for GM-CSF: Results in vitro, in rodents and in humans. Int. J. Cancer 2015, 137, 1775–1783.
- Kloos, A.; Woller, N.; Gürlevik, E.; Ureche, Cr.; Niemann, J.; Armbrecht, N.; Martin, N.T.; Geffers, R.; Manns, M.P.; Gerardy-Schahn, R. PolySia-Specific Retargeting of Oncolytic Viruses Triggers Tumor-Specific Immune Responses and Facilitates Therapy of Disseminated Lung Cancer. Cancer Immunol. Res. 2015, 3, 751–763.Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17.
- Francisco, J.A.; Gilliland, L.K.; Stebbins, M.R.; Norris, N.A.; Ledbetter, J.A.; Siegall, C.B. Activity of a single-chain immunotoxin that selectively kills lymphoma and other B-lineage cells expressing the CD40 antigen. Cancer Res. 1995, 55, 3099–3104.Salzwedel, A.O.; Han, J.; LaRocca, C.J.; Shanley, R.; Yamamoto, M.; Davydova, J. Combination of interferon-expressing oncolytic adenovirus with chemotherapy and radiation is highly synergistic in hamster model of pancreatic cancer. Oncotarget 2018, 9, 18041–18052.
- Chen, C.Y.; May, S.M.; Barry, M.A. Targeting Adenoviruses Factor X-Single-Chain Antibody Fusion Proteins. Hum. Gene Ther. 2010, 21, 739–749.Armstrong, L.; Arrington, A.; Han, J.; Gavrikova, T.; Brown, E.; Yamamoto, M.; Vickers, S.M.; Davydova, J. Generation of a novel, cyclooxygenase-2-targeted, interferon-expressing, conditionally replicative adenovirus for pancreatic cancer therapy. Am. J. Surg. 2012, 204, 741–750.
- Garas, M.N.; Tillib, S.V.; Zubkova, O.V.; Rogozhin, V.N.; Ivanova, T.I.; Vasilev, L.A.; Logunov, D.Y.; Shmarov, M.M.; Tutykhina, I.L.; Esmagambetov, I.B. Construction of a pIX-modified Adenovirus Vector Able to Effectively Bind to Nanoantibodies for Targeting. Acta Nat. 2014, 6, 95–105.Armstrong, L.; Davydova, J.; Brown, E.; Han, J.; Yamamoto, M.; Vickers, S.M. Delivery of interferon alpha using a novel Cox2-controlled adenovirus for pancreatic cancer therapy. Surgery 2012, 152, 114–122.
- Wang, Z.; Yu, B.; Wang, B.; Yan, J.; Feng, X.; Wang, Z.; Wang, L.; Zhang, H.; Wu, H.; Wu, J. A novel capsid-modified oncolytic recombinant adenovirus type 5 for tumor-targeting gene therapy by intravenous route. Oncotarget 2016, 7, 47287–47301.Boettcher, M.; McManus, M.T. Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585.
- Glasgow, J.N.; Mikheeva, G.; Krasnykh, V.; Curiel, D.T. A strategy for adenovirus vector targeting with a secreted single chain antibody. PLoS ONE 2009, 4, e8355.Singh, S.K.; Bhadra, U.; Hajeri, P.B.; Pal-Bhadra, M. MicroRNA Tales in Fly Development. In Regulation of Gene Expression by Small RNAs; Gaur, R.K., Rossi, J.J., Eds.; CRC Press: Boca Raton, FL, USA, 2009; pp. 123–147.
- Jakubczak, J.L.; Rollence, M.L.; Stewart, D.A.; Jafari, J.D.; von Seggern, D.J.; Nemerow, G.R.; Stevenson, S.C.; Hallenbeck, P.L. Adenovirus type 5 viral particles pseudotyped with mutagenized fiber proteins show diminished infectivity of coxsackie B-adenovirus receptor-bearing cells. J. Virol. 2001, 75, 2972–2981.Singh, S.K.; Hajeri, P.B. Rna Interference: From basics to Therapeutics. In Molecular and Cellular Therapeutics; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2012; pp. 140–167.
- Porter, C.E.; Shaw, A.R.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262.Azlan, A.; Dzaki, N.; Azzam, G. Argonaute: The executor of small RNA function. J. Genet. Genom. 2016, 43, 481–494.
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; jan Krönke; Facon, T.; Salnikov, A.V.; Lesley, R.; Beutner, K. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783.Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108.
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Jr.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616.Wu, S.Y.; Lopez-Berestein, G.; Calin, G.A.; Sood, A.K. RNAi therapies: Drugging the undruggable. Sci. Transl. Med. 2014, 6, 240ps7.
- Freedman, J.D.; Hagel, J.; Scott, E.M.; Psallidas, I.; Gupta, A.; Spiers, L.; Miller, P.; Kanellakis, N.; Ashfield, R.; Fisher, K.D. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol. Med. 2017, 9, 1067–1087.Deng, Y.; Wang, C.C.; Choy, K.W.; Du, Q.; Chen, J.; Wang, Q.; Li, L.; Chung, T.K.H.; Tang, T. Therapeutic potentials of gene silencing by RNA interference: Principles, challenges, and new strategies. Gene 2014, 538, 217–227.
- Fajardo, C.A.; Guedan, S.; Rojas, L.A.; Moreno, R.; Arias-Badia, M.; de Sostoa, J.; June, C.H.; Alemany, R. Oncolytic Adenoviral Delivery of an EGFR-Targeting T-cell Engager Improves Antitumor Efficacy. Cancer Res. 2017, 77, 2052–2063.Hajeri, P.B.; Singh, S.K. siRNAs: Their potential as therapeutic agents—Part I. Designing of siRNAs. Drug Discov. Today 2009, 14, 851–858.
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865.Singh, S.K.; Hajeri, P.B. siRNAs: Their potential as therapeutic agents—Part II. Methods of delivery. Drug Discov. Today 2009, 14, 859–865.
- Bramante, S.; Kaufmann, J.K.; Veckman, V.; Liikanen, I.; Nettelbeck, D.M.; Hemminki, O.; Vassilev, L.; Cerullo, V.; Oksanen, M.; Heiskanen, R., et al. Treatment of melanoma with a serotype 5/3 chimeric oncolytic adenovirus coding for GM-CSF: Results in vitro, in rodents and in humans. International journal of cancer 2015, 137, 1775-1783, doi:10.1002/ijc.29536.Buduru, S.; Zimta, A.A.; Ciocan, C.; Braicu, C.; Dudea, D.; Irimie, A.I.; Berindan-Neagoe, I. RNA interference: New mechanistic and biochemical insights with application in oral cancer therapy. Int. J. Nanomed. 2018, 13, 3397–3409.
- Krasnykh, V.N.; Mikheeva, G.V.; Douglas, J.T.; Curiel, D.T. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J. Virol. 1996, 70, 6839–6846.Liu, Y.P.; Berkhout, B. miRNA cassettes in viral vectors: Problems and solutions. Biochim. Biophys. Acta 2011, 1809, 732–745.
- Dranoff, G. GM-CSF-secreting melanoma vaccines. Oncogene 2003, 22, 3188–3192.Pei, D.S.; Di, J.H.; Chen, F.F.; Zheng, J.N. Oncolytic-adenovirus-expressed RNA interference for cancer therapy. Expert Opin. Biol. Ther. 2010, 10, 1331–1341.
- Ramesh, N.; Ge, Y.; Ennist, D.L.; Zhu, M.; Mina, M.; Ganesh, S.; Reddy, P.S.; Yu, D.-C. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor--armed oncolytic adenovirus for the treatment of bladder cancer. Clin. Cancer. Res. 2006, 12, 305–313.Li, Y.; Zhang, B.; Zhang, H.; Zhu, X.; Feng, D.; Zhang, D.; Zhuo, B.; Li, L.; Zheng, J. Oncolytic adenovirus armed with shRNA targeting MYCN gene inhibits neuroblastoma cell proliferation and in vivo xenograft tumor growth. J. Cancer. Res. Clin. Oncol. 2013, 139, 933–941.
- Flemington, E.K.; Speck, S.H.; Kaelin, W.G.; Jr. E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc. Natl. Acad. Sci. USA 1993, 90, 6914–6918.Lu, S.; Cullen, B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004, 78, 12868–12876.
- Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J. Urol. 2012, 188, 2391–2397.Yang, Z.; Li, Y. Dissection of RNAi-based antiviral immunity in plants. Curr. Opin. Virol. 2018, 32, 88–99.
- Gregory, S.M.; Nazir, S.A.; Metcalf, J.P. Implications of the innate immune response to adenovirus and adenoviral vectors. Future Virol. 2011, 6, 357–374.Schuster, S.; Miesen, P.; van Rij, R.P. Antiviral RNAi in Insects and Mammals: Parallels and Differences. Viruses 2019, 11, 448.
- Tong, A.W.; Stone, M.J. Prospects for Cd40-directed experimental therapy of human cancer. Cancer Gene Ther. 2003, 10, 1–13.Zhao, J.H.; Hua, C.L.; Fang, Y.Y.; Guo, H.S. The dual edge of RNA silencing suppressors in the virus-host interactions. Curr. Opin. Virol. 2016, 17, 39–44.
- Tong, A.W.; Papayoti, M.H.; Netto, G.; Armstrong, D.T.; Ordonez, G.; Lawson, J.M.; Stone, M.J. Growth-inhibitory effects of CD40 ligand (CD154) and its endogenous expression in human breast cancer. Clin. Cancer Res. 2001, 7, 691–703.Machitani, M.; Sakurai, F.; Katayama, K.; Tachibana, M.; Suzuki, T.; Matsui, H.; Yamaguchi, T.; Mizuguchi, H. Improving adenovirus vector-mediated RNAi efficiency by lacking the expression of virus-associated RNAs. Virus Res. 2013, 178, 357–363.
- Ghamande, S.; Hylander, B.L.; Oflazoglu, E.; Lele, S.; Fanslow, W.; Repasky, E.A. Recombinant CD40 ligand therapy has significant antitumor effects on CD40-positive ovarian tumor xenografts grown in SCID mice and demonstrates an augmented effect with cisplatin. Cancer Res. 2001, 61, 7556–7562.Aparicio, O.; Razquin, N.; Zaratiegui, M.; Narvaiza, I.; Fortes, P. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J. Virol. 2006, 80, 1376–1384.
- Nukui, Y.; Picozzi, V.J.; Traverso, L.W. Interferon-based adjuvant chemoradiation therapy improves survival after pancreaticoduodenectomy for pancreatic adenocarcinoma. Am. J. Surg. 2000, 179, 367–371.Bennasser, Y.; Chable-Bessia, C.; Triboulet, R.; Gibbings, D.; Gwizdek, C.; Dargemont, C.; Kremer, E.J.; Voinnet, O.; Benkirane, M. Competition for XPO5 binding between Dicer mRNA, pre-miRNA and viral RNA regulates human Dicer levels. Nat. Struct. Mol. Biol. 2011, 18, 323–327.
- Ohashi, M.; Yoshida, K.; Kushida, M.; Miura, Y.; Ohnami, S.; Ikarashi, Y.; Kitade, Y.; Yoshida, T.; Aoki, K. Adenovirus-mediated interferon alpha gene transfer induces regional direct cytotoxicity and possible systemic immunity against pancreatic cancer. Br. J. Cancer 2005, 93, 441–449.Machitani, M.; Yamaguchi, T.; Shimizu, K.; Sakurai, F.; Katayama, K.; Kawabata, K.; Mizuguchi, H. Adenovirus Vector-Derived VA-RNA-Mediated Innate Immune Responses. Pharmaceutics 2011, 3, 338–353.
- Hara, H.; Kobayashi, A.; Yoshida, K.; Ohashi, M.; Ohnami, S.; Uchida, E.; Higashihara, E.; Yoshida, T.; Aoki, K. Local interferon-alpha gene therapy elicits systemic immunity in a syngeneic pancreatic cancer model in hamster. Cancer Sci. 2007, 98, 455–463.Kitajewski, J.; Schneider, R.J.; Safer, B.; Munemitsu, S.M.; Samuel, C.E.; Thimmappaya, B.; Shenk, T. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 1986, 45, 195–200.
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Löskog, A. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998.Vachon, V.K.; Conn, G.L. Adenovirus VA RNA: An essential pro-viral non-coding RNA. Virus Res. 2016, 212, 39–52.
- Li, S.; Qi, Z.; Li, H.; Hu, J.; Wang, D.; Wang, X.; Feng, Z. Conditionally replicating oncolytic adenoviral vector expressing arresten and tumor necrosis factor-related apoptosis-inducing ligand experimentally suppresses lung carcinoma progression. Mol. Med. Rep. 2015, 12, 2068–2074.Carette, J.E.; Overmeer, R.M.; Schagen, F.H.; Alemany, R.; Barski, O.A.; Gerritsen, W.R.; Van Beusechem, V.W. Conditionally replicating adenoviruses expressing short hairpin RNAs silence the expression of a target gene in cancer cells. Cancer Res. 2004, 64, 2663–2667.
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17.Rovira-Rigau, M.; Raimondi, G.; Marín, M.Á.; Gironella, M.; Alemany, R.; Fillat, C. Bioselection Reveals miR-99b and miR-485 as Enhancers of Adenoviral Oncolysis in Pancreatic Cancer. Mol. Ther. 2019, 27, 230–243.
- Salzwedel, A.O.; Han, J.; LaRocca, C.J.; Shanley, R.; Yamamoto, M.; Davydova, J. Combination of interferon-expressing oncolytic adenovirus with chemotherapy and radiation is highly synergistic in hamster model of pancreatic cancer. Oncotarget 2018, 9, 18041–18052.Brachtlova, T.; van Beusechem, V.W. Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens. Cells 2018, 7, 288.
- Armstrong, L.; Arrington, A.; Han, J.; Gavrikova, T.; Brown, E.; Yamamoto, M.; Vickers, S.M.; Davydova, J. Generation of a novel, cyclooxygenase-2-targeted, interferon-expressing, conditionally replicative adenovirus for pancreatic cancer therapy. Am. J. Surg. 2012, 204, 741–750.Machitani, M.; Sakurai, F.; Wakabayashi, K.; Tachibana, M.; Fujiwara, T.; Mizuguchi, H. Enhanced Oncolytic Activities of the Telomerase-Specific Replication-Competent Adenovirus Expressing Short-Hairpin RNA against Dicer. Mol. Cancer Ther. 2017, 16, 251–259.
- Armstrong, L.; Davydova, J.; Brown, E.; Han, J.; Yamamoto, M.; Vickers, S.M. Delivery of interferon alpha using a novel Cox2-controlled adenovirus for pancreatic cancer therapy. Surgery 2012, 152, 114–122.Luo, Q.; Song, H.; Deng, X.; Li, J.; Jian, W.; Zhao, J.; Zheng, X.; Basnet, S.; Ge, H.; Daniel, T. A Triple-Regulated Oncolytic Adenovirus Carrying MicroRNA-143 Exhibits Potent Antitumor Efficacy in Colorectal Cancer. Mol. Ther. Oncolytics 2020, 16, 219–229.
- Boettcher, M. and M.T. McManus, Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585.Callegari, E.; Elamin, B.K.; D’Abundo, L.; Falzoni, S.; Donvito, G.; Moshiri, F.; Milazzo, M.; Altavilla, G.; Giacomelli, L.; Fornari, F. Anti-tumor activity of a miR-199-dependent oncolytic adenovirus. PLoS ONE 2013, 8, e73964.
- Singh, S.K.; Bhadra, U.; Hajeri, P.B.; Pal-Bhadra, M. MicroRNA Tales in Fly Development. In Regulation of Gene Expression by Small RNAs; Gaur, R.K., Rossi, J.J., Eds.; CRC Press: Boca Raton, FL, USA, 2009; pp. 123–147.Ang, L.; Guo, L.; Wang, J.; Huang, J.; Lou, X.; Zhao, M. Oncolytic virotherapy armed with an engineered interfering lncRNA exhibits antitumor activity by blocking the epithelial mesenchymal transition in triple-negative breast cancer. Cancer Lett. 2020, 49, 42–53.
- Hajeri, P.B.; Singh, S.K. Rna Interference: From basics to Therapeutics. In Molecular and Cellular Therapeutics; Whitehouse, D., Rapley, R., Eds.; Wiley: Hoboken, NJ, USA, 2012; pp. 140–167.Tang, S.; Tan, G.; Jiang, X.; Han, P.; Zhai, B.; Dong, X.; Qiao, H.; Jiang, H.; Sun, X. An artificial lncRNA targeting multiple miRNAs overcomes sorafenib resistance in hepatocellular carcinoma cells. Oncotarget 2016, 7, 73257–73269.
- Azlan, A.; Dzaki, N.; Azzam, G. Argonaute: The executor of small RNA function. J. Genet. Genom. 2016, 43, 481–494.Luo, Q.; Basnet, S.; Dai, Z.; Li, S.; Zhang, Z.; Ge, H. A novel E1B55kDa-deleted oncolytic adenovirus carrying microRNA-143 exerts specific antitumor efficacy on colorectal cancer cells. Am. J. Transl. Res. 2016, 8, 3822–3830.
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108.Wang, M.; Liu, J.; Xi, D.; Luo, X.; Ning, Q. Adenovirus-mediated artificial microRNA against human fibrinogen like protein 2 inhibits hepatocellular carcinoma growth. J. Gene Med. 2016, 18, 102–111.
- Wu, S.Y.; Lopez-Berestein, G.; Calin, G.A.; Sood, A.K. RNAi therapies: Drugging the undruggable. Sci. Transl. Med. 2014, 6, 240ps7.Lou, W.; Chen, Q.; Ma, L.; Liu, J.; Yang, Z.; Shen, J.; Cui, Y.; Bian, X.; Qian, C. Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. J. Mol. Med. 2013, 91, 715–725.
- Deng, Y.; Wang, C.C.; Choy, K.W.; Du, Q.; Chen, J.; Wang, Q.; Li, L.; Chung, T.K.H.; Tang, T. Therapeutic potentials of gene silencing by RNA interference: Principles, challenges, and new strategies. Gene 2014, 538, 217–227.Kosaka, T.; Davydova, J.; Ono, H.A.; Akiyama, H.; Hirai, S.; Ohno, S.; Takeshita, F.; Aoki, K.; Ochiya, T.; Yamamoto, M.; et al. Imaging and Antitumoral Effect of a Cyclo-oxygenase 2-specific Replicative Adenovirus for Small Metastatic Gastric Cancer Lesions. Anticancer Res. 2015, 35, 5201–5210.
- Hajeri, P.B.; Singh, S.K. siRNAs: Their potential as therapeutic agents--Part, I. Designing of siRNAs. Drug Discov. Today 2009, 14, 851–858.LaRocca, C.J.; Han, J.; Salzwedel, A.O.; Davydova, J.; Herzberg, M.C.; Gopalakrishnan, R.; Yamamoto, M. Oncolytic adenoviruses targeted to Human Papilloma Virus-positive head and neck squamous cell carcinomas. Oral Oncol. 2016, 56, 25–31.
- Singh, S.K.; Hajeri, P.B. siRNAs: Their potential as therapeutic agents--Part II. Methods of delivery. Drug Discov. Today 2009, 14, 859–865.Yamamoto, M.; Davydova, J.; Wang, M.; Siegal, G.P.; Krasnykh, V.; Vickers, S.M.; Curiel, D.T. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology 2003, 125, 1203–1218.
- Buduru, S.; Zimta, A.A.; Ciocan, C.; Braicu, C.; Dudea, D.; Irimie, A.I.; Berindan-Neagoe, I. RNA interference: New mechanistic and biochemical insights with application in oral cancer therapy. Int. J. Nanomed. 2018, 13, 3397–3409.Suzuki, S.; Kofune, H.; Uozumi, K.; Yoshimitsu, M.; Arima, N.; Ishitsuka, K.; Ueno, S.-I.; Kosa, K.-I. A survivin-responsive, conditionally replicating adenovirus induces potent cytocidal effects in adult T-cell leukemia/lymphoma. BMC Cancer 2019, 19, 516.
- Liu, Y.P.; Berkhout, B.; miRNA cassettes in viral vectors: Problems and solutions. Biochim Biophys Acta 2011, 1809, 732–745.Ali, S.; Tahir, M.; Khan, A.A.; Chen, X.C.; Ling, M.; Huan, Y. Cisplatin Synergistically Enhances Antitumor Potency of Conditionally Replicating Adenovirus via p53 Dependent or Independent Pathways in Human Lung Carcinoma. Int. J. Mol. Sci. 2019, 20, 1125.
- Pei, D.S.; Di, J.H.; Chen, F.F.; Zheng, J.N. Oncolytic-adenovirus-expressed RNA interference for cancer therapy. Expert Opin. Biol. Ther. 2010, 10, 1331–1341.Mooney, R.; Majid, A.A.; Batalla-Covello, J.; Machad, D.; Liu, X.; Gonzaga, J.; Tirughana, R.; Hammad, M.; Lesniak, M.S.; Curiel, D.T. Enhanced Delivery of Oncolytic Adenovirus by Neural Stem Cells for Treatment of Metastatic Ovarian Cancer. Mol. Ther. Oncolytics 2019, 12, 79–92.
- Li, Y.; Zhang, B.; Zhang, H.; Zhu, X.; Feng, D.; Zhang, D.; Zhuo, B.; Li, L.; Zheng, J. Oncolytic adenovirus armed with shRNA targeting MYCN gene inhibits neuroblastoma cell proliferation and in vivo xenograft tumor growth. J. Cancer. Res. Clin. Oncol. 2013, 139, 933–941.Yamamoto, Y.; Nagasato, M.; Rin, Y.; Henmi, M.; Ino, Y.; Yachida, S.; Ohki, R.; Hiraoka, N.; Tagawa, M.; Aoki, K. Strong antitumor efficacy of a pancreatic tumor-targeting oncolytic adenovirus for neuroendocrine tumors. Cancer Med. 2017, 6, 2385–2397.
- Callegari, E.; Elamin, B.K.; D'Abundo, L.; Falzoni, S.; Donvito, G.; Moshiri, F.; Milazzo, M.; Altavilla, G.; Giacomelli, L.; Fornari, F. Anti-tumor activity of a miR-199-dependent oncolytic adenovirus. PLoS ONE 2013, 8, e73964.Cherry, T.; Longo, S.L.; Tovar-Spinoza, Z.; Post, D.E. Second-generation HIF-activated oncolytic adenoviruses with improved replication, oncolytic, and antitumor efficacy. Gene Ther. 2010, 17, 1430–1441.
- Ang, L.; Guo, L.; Wang, J.; Huang, J.; Lou, X.; Zhao, M. Oncolytic virotherapy armed with an engineered interfering lncRNA exhibits antitumor activity by blocking the epithelial mesenchymal transition in triple-negative breast cancer. Cancer Lett. 2020, 49, 42–53.Post, D.E.; Sandberg, E.M.; Kyle, M.M.; Devi, N.S.; Brat, D.J.; Xu, Z.; Tighiouart, M.; van Meir, E.G. Targeted cancer gene therapy using a hypoxia inducible factor dependent oncolytic adenovirus armed with interleukin-4. Cancer Res. 2007, 67, 6872–6881.
- Tang, S.; Tan, G.; Jiang, X.; Han, P.; Zhai, B.; Dong, X.; Qiao, H.; Jiang, H.; Sun, X. An artificial lncRNA targeting multiple miRNAs overcomes sorafenib resistance in hepatocellular carcinoma cells. Oncotarget 2016, 7, 73257–73269.Tazawa, H.; Hasei, J.; Yano, S.; Kagawa, S.; Ozaki, T.; Fujiwara, T. Bone and Soft-Tissue Sarcoma: A New Target for Telomerase-Specific Oncolytic Virotherapy. Cancers 2020, 12, 478.
- Luo, Q.; Basnet, S.; Dai, Z.; Li, S.; Zhang, Z.; Ge, H. A novel E1B55kDa-deleted oncolytic adenovirus carrying microRNA-143 exerts specific antitumor efficacy on colorectal cancer cells. Am. J. Transl. Res. 2016, 8, 3822–3830.Cui, C.X.; Li, Y.-Q.; Sun, Y.-J.; Zhu, Y.-L.; Fang, J.-B.; Bai, B.; Li, W.-J.; Li, S.-Z.; Ma, Y.-Z.; Li, X. Antitumor effect of a dual cancer-specific oncolytic adenovirus on prostate cancer PC-3 cells. Urol. Oncol. 2019, 37, 352.e1–352.e18.
- Wang, M.; Liu, J.; Xi, D.; Luo, X.; Ning, Q. Adenovirus-mediated artificial microRNA against human fibrinogen like protein 2 inhibits hepatocellular carcinoma growth. J. Gene Med. 2016, 18, 102–111.Lin, J.; Zeng, D.; He, H.; Tan, G.; Lan, Y.; Jiang, F.; Sheng, S. Gene therapy for human ovarian cancer cells using efficient expression of Fas gene combined with gammadeltaT cells. Mol. Med. Rep. 2017, 16, 3791–3798.
- Lou, W.; Chen, Q.; Ma, L.; Liu, J.; Yang, Z.; Shen, J.; Cui, Y.; Bian, Xi.; Qian, C. Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. J. Mol. Med. 2013, 91, 715–725.Kim, Y.H.; Kim, K.T.; Lee, S.A.; Hong, S.E.; Moon, J.Y.; Yoon, E.K.; Kim, S.; Kim, E.O.; Kang, S.H.; Kim, S.K. Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics 2016, 6, 357–368.
- Luo, Q.; Song, H.; Deng, X.; Li, J.; Jian, W.; Zhao, J.; Zheng, X.; Basnet, S.; Ge, H.; Daniel, T. A Triple-Regulated Oncolytic Adenovirus Carrying MicroRNA-143 Exhibits Potent Antitumor Efficacy in Colorectal Cancer. Mol. Ther. Oncolytics 2020, 16, 219–229.Wu, X.; Chen, J.; Cao, Y.; Xie, B.; Li, H.; Zhou, P.; Qiu, Y.; Pang, J. Antitumor effect of COOH-terminal polypeptide of human TERT is associated with the declined expression of hTERT and NF-kappaB p65 in HeLa cells. Oncol. Rep. 2015, 34, 2909–2916.
- Kosaka, T.; Davydova, J.; Ono, H.A.; Akiyama, H.; Hirai, Sh.; Ohno, S.; Takeshita, F.; Aoki, K.; Ochiya, T.; Yamamoto, M.; Kunisaki, C. Imaging and Antitumoral Effect of a Cyclo-oxygenase 2-specific Replicative Adenovirus for Small Metastatic Gastric Cancer Lesions. Anticancer Res. 2015, 35, 5201–5210.Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 2000, 6, 1134–1139.
- LaRocca, C.J.; Han, J.; Salzwedel, A.O.; Davydova, J.; Herzberg, M.C.; Gopalakrishnan, R.; Yamamoto, M. Oncolytic adenoviruses targeted to Human Papilloma Virus-positive head and neck squamous cell carcinomas. Oral Oncol. 2016, 56, 25–31.Heise, C.; Sampson-Johannes, A.; Williams, A.; Mccormick, F.; Von Hoff, D.D.; Kirn, D.H. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997, 3, 639–645.
- Yamamoto, M.; Davydova, J.; Wang, M.; Siegal, G.P.; Krasnykh, V.; Vickers, S.M.; Curiel, D.T. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology 2003, 125, 1203–1218.Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12.
- Suzuki, S.; Kofune, H.; Uozumi, K.; Yoshimitsu, M.; Arima, N.; Ishitsuka, K.; Ueno, S.-I.; Kosa, K.-I. A survivin-responsive, conditionally replicating adenovirus induces potent cytocidal effects in adult T-cell leukemia/lymphoma. BMC Cancer 2019, 19, 516.Ganesh, S.; Gonzalez-Edick, M.; Gibbons, D.; Ge, Y.; VanRoey, M.; Robinson, M.; Jooss, K. Combination therapy with radiation or cisplatin enhances the potency of Ad5/35 chimeric oncolytic adenovirus in a preclinical model of head and neck cancer. Cancer Gene Ther. 2009, 16, 383–392.
- Ali, S.; Tahir, M.; Khan, A.A.; Chen, X.C.; Ling, M.; Huan, Y. Cisplatin Synergistically Enhances Antitumor Potency of Conditionally Replicating Adenovirus via p53 Dependent or Independent Pathways in Human Lung Carcinoma. Int. J. Mol. Sci. 2019, 20, 1125.Reid, T.R.; Freeman, S.; Post, L.; McCormick, F.; Sze, D.Y. Effects of Onyx-015 among metastatic colorectal cancer patients that have failed prior treatment with 5-FU/leucovorin. Cancer Gene Ther. 2005, 12, 673–681.
- Mooney, R.; Majid, A.A.; Batalla-Covello, J.; Machad, D.; Liu, X.; Gonzaga, J.; Tirughana, R.; Hammad, M.; Lesniak, M.S.; Curiel, D.T. Enhanced Delivery of Oncolytic Adenovirus by Neural Stem Cells for Treatment of Metastatic Ovarian Cancer. Mol. Ther. Oncolytics 2019, 12, 79–92.Graat, H.C.; Carette, J.E.; Schagen, F.H.E.; Vassilev, L.T.; Gerritsen, W.R.; Kaspers, G.J.L.; Wuisman, P.I.J.M.; van Beusechem, V.W. Enhanced tumor cell kill by combined treatment with a small-molecule antagonist of mouse double minute 2 and adenoviruses encoding p53. Mol. Cancer Ther. 2007, 6, 1552–1561.
- Yamamoto, Y.; Nagasato, M.; Rin, Y.; Henmi, M.; Ino, Y.; Yachida, S.; Ohki, R.; Hiraoka, N.; Tagawa, M.; Aoki, K. Strong antitumor efficacy of a pancreatic tumor-targeting oncolytic adenovirus for neuroendocrine tumors. Cancer Med. 2017, 6, 2385–2397.Tam, C.; Tam, C.; Wong, J.H.; Tsui, S.K.W.; Zuo, T.; Chan, T.F.; Ng, T.B. LncRNAs with miRNAs in regulation of gastric, liver, and colorectal cancers: Updates in recent years. Appl. Microbiol. Biotechnol. 2019, 103, 4649–4677.
- Tam, C.; Tam, C.; Wong, J.H.; Tsui, S.K.W.; Zuo, T.; Chan, T.F.; Ng, T.B. LncRNAs with miRNAs in regulation of gastric, liver, and colorectal cancers: Updates in recent years. Appl. Microbiol. Biotechnol. 2019, 103, 4649–4677.Shayestehpour, M.; Moghim, S.; Salimi, V.; Jalilvand, S.; Yavarian, J.; Romani, B.; Mokhtari-Azad, T. Targeting human breast cancer cells by an oncolytic adenovirus using microRNA-targeting strategy. Virus Res. 2017, 240, 207–214.
- Shayestehpour, M.; Moghim, S.; Salimi, V.; Jalilvand, S.; Yavarian, J.; Romani, B.; Mokhtari-Azad, T. Targeting human breast cancer cells by an oncolytic adenovirus using microRNA-targeting strategy. Virus Res. 2017, 240, 207–214.Bofill-De Ros, X.; Gironella, M.; Fillat, C. miR-148a- and miR-216a-regulated oncolytic adenoviruses targeting pancreatic tumors attenuate tissue damage without perturbation of miRNA activity. Mol. Ther. 2014, 22, 1665–1677.
- Bofill-De Ros, X.; Gironella, M.; Fillat, C. miR-148a- and miR-216a-regulated oncolytic adenoviruses targeting pancreatic tumors attenuate tissue damage without perturbation of miRNA activity. Mol. Ther. 2014, 22, 1665–1677.Zhang, Z.; Zhang, X.; Newman, K.; Liu, X. MicroRNA regulation of oncolytic adenovirus 6 for selective treatment of castration-resistant prostate cancer. Mol. Cancer Ther. 2012, 11, 2410–2418.
- Zhang, Z.; Zhang, X.; Newman, K.; Liu, X. MicroRNA regulation of oncolytic adenovirus 6 for selective treatment of castration-resistant prostate cancer. Mol. Cancer Ther. 2012, 11, 2410–2418.Bofill-De Ros, X.; Villanueva, E.; Fillat, C. Late-phase miRNA-controlled oncolytic adenovirus for selective killing of cancer cells. Oncotarget 2015, 6, 6179–6190.
- Bofill-De Ros, X.; Villanueva, E.; Fillat, C. Late-phase miRNA-controlled oncolytic adenovirus for selective killing of cancer cells. Oncotarget 2015, 6, 6179–6190.Wang, X.W.; Zhang, C.; Lee, K.C.; He, X.J.; Lu, Z.Q.; Huang, C.; Wu, Q.C. Adenovirus-Mediated Gene Transfer of microRNA-21 Sponge Inhibits Neointimal Hyperplasia in Rat Vein Grafts. Int. J. Biol. Sci. 2017, 13, 1309–1319.
- Wang, X.W.; Zhang, C.; Lee, K.C.; He, X.J.; Lu, Z.Q.; Huang, C.; Wu, Q.C. Adenovirus-Mediated Gene Transfer of microRNA-21 Sponge Inhibits Neointimal Hyperplasia in Rat Vein Grafts. Int. J. Biol. Sci. 2017, 13, 1309–1319.Jin, H.; Lv, S.; Yang, J.; Wang, X.; Hu, H.; Su, C.; Zhou, C.; Li, J.; Huang, Y.; Li, L. Use of microRNA Let-7 to control the replication specificity of oncolytic adenovirus in hepatocellular carcinoma cells. PLoS ONE 2011, 6, e21307.
- Jin, H.; Lv, S.; Yang, J.; Wang, X.; Hu, H.; Su, C.; Zhou, C.; Li, J.; Huang, Y.; Li, L. Use of microRNA Let-7 to control the replication specificity of oncolytic adenovirus in hepatocellular carcinoma cells. PLoS ONE 2011, 6, e21307.Shayestehpour, M.; Moghim, S.; Salimi, V.; Jalilvand, S.; Yavarian, J.; Romani, B.; Ylosmaki, E.; Mokhtari-Azad, T. Selective replication of miR-145-regulated oncolytic adenovirus in MCF-7 breast cancer cells. Future Virol. 2016, 11, 671–680.
- Shayestehpour, M.; Moghim, S.; Salimi, V.; Jalilvand, S.; Yavarian, J.; Romani, B.; Ylosmaki, E.; Mokhtari-Azad, T. Selective replication of miR-145-regulated oncolytic adenovirus in MCF-7 breast cancer cells. Future Virol. 2016, 11, 671–680.Leja, J.; Nilsson, B.; Di, Y.; Gustafson, E.; Åkerström, G.; Öberg, K.; Giandomenico, V.; Essand, M. Double-detargeted oncolytic adenovirus shows replication arrest in liver cells and retains neuroendocrine cell killing ability. PLoS ONE 2010, 5, e8916.
- Leja, J.; Nilsson, B.; Di, Y.; Gustafson, E.; Åkerström, G.; Öberg, K.; Giandomenico, V.; Essand, M. Double-detargeted oncolytic adenovirus shows replication arrest in liver cells and retains neuroendocrine cell killing ability. PLoS ONE 2010, 5, e8916.Kabekkodu, S.P.; Shukla, V.; Varghese, V.K.; Adiga, D.; Vethil, J.P.; Chakrabarty, S.; Satyamoorthy, K. Cluster miRNAs and cancer: Diagnostic, prognostic and therapeutic opportunities. Wiley Interdiscip. Rev. RNA 2020, 11, e1563.
- Kabekkodu, S.P.; Shukla, V.; Varghese, V.K.; Adiga, D.; Vethil, J.P.; Chakrabarty, S.; Satyamoorthy, K. Cluster miRNAs and cancer: Diagnostic, prognostic and therapeutic opportunities. Wiley Interdiscip. Rev. RNA 2020, 11, e1563.Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399.
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399.Karimian, A.; Azizian, K.; Parsian, H.; Rafieian, S.; Shafiei-Irannejad, V.; Kheyrollah, M.; Yousefi, M.; Majidinia, M.; Yousefi, B. CRISPR/Cas9 technology as a potent molecular tool for gene therapy. J. Cell Physiol. 2019, 234, 12267–12277.
- Karimian, A.; Azizian, K.; Parsian, H.; Rafieian, S.; Shafiei-Irannejad, V.; Kheyrollah, M.; Yousefi, M.; Majidinia, M.; Yousefi, B. CRISPR/Cas9 technology as a potent molecular tool for gene therapy. J. Cell Physiol. 2019, 234, 12267-12277.Chen, M.; Mao, A.; Xu, M.; Weng, Q.; Mao, J.; Ji, J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019, 447, 48–55.
- Chen, M.; Mao, A.; Xu, M.; Weng, Q.; Mao, J.; Ji, J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019, 447, 48–55.Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105.
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105.Holkers, M.; Maggio, I.; Liu, J.; Janssen, J.M.; Miselli, F.; Mussolino, C.; Recchia, A.; Cathomen, T.; Gonçalves, M.A.F.V. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013, 41, e63.
- Holkers, M.; Maggio, I.; Liu, J.; Janssen, J.M.; Miselli, F.; Mussolino, C.; Recchia, A.; Cathomen, T.; Gonçalves, M.A.F.V. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013, 41, e63.Maggio, I.; Holkers, M.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A. Adenoviral vector delivery of RNA-guided CRISPR/Cas9 nuclease complexes induces targeted mutagenesis in a diverse array of human cells. Sci. Rep. 2014, 4, 5105.
- Maggio, I.; Holkers, M.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A. Adenoviral vector delivery of RNA-guided CRISPR/Cas9 nuclease complexes induces targeted mutagenesis in a diverse array of human cells. Sci. Rep. 2014, 4, 5105.Palmer, D.J.; Turner, D.L.; Ng, P. Production of CRISPR/Cas9-Mediated Self-Cleaving Helper-Dependent Adenoviruses. Mol. Ther. Methods Clin. Dev. 2019, 13, 432–439.
- Palmer, D.J.; Turner, D.L.; Ng, P. Production of CRISPR/Cas9-Mediated Self-Cleaving Helper-Dependent Adenoviruses. Mol. Ther. Methods Clin. Dev. 2019, 13, 432–439.Li, C.; Psatha, N.; Gil, S.; Wang, H.; Papayannopoulou, T.; Lieber, A. HDAd5/35(++) Adenovirus Vector Expressing Anti-CRISPR Peptides Decreases CRISPR/Cas9 Toxicity in Human Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev. 2018, 9, 390–401.
- Li, C.; Psatha, N.; Gil, S.; Wang, H.; Papayannopoulou, T.; Lieber, A. HDAd5/35(++) Adenovirus Vector Expressing Anti-CRISPR Peptides Decreases CRISPR/Cas9 Toxicity in Human Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev. 2018, 9, 390–401.Palmer, D.J.; Turner, D.L.; Ng, P. A Single “All-in-One” Helper-Dependent Adenovirus to Deliver Donor DNA and CRISPR/Cas9 for Efficient Homology-Directed Repair. Mol. Ther. Methods. Clin. Dev. 2020, 17, 441–447.
- Palmer, D.J.; Turner, D.L.; Ng, P. A Single “All-in-One” Helper-Dependent Adenovirus to Deliver Donor DNA and CRISPR/Cas9 for Efficient Homology-Directed Repair. Mol. Ther. Methods. Clin. Dev. 2020, 17, 441–447.Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119.
- Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119.Ashmore-Harris, C.; Fruhwirth, G.O. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin. Transl. Med. 2020, 9, 15.
- Ashmore-Harris, C.; Fruhwirth, G.O. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin. Transl. Med. 2020, 9, 15.Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target Ther. 2020, 5, 1.
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct Target Ther. 2020, 5, 1.Li, C.; Lieber, A. Adenovirus vectors in hematopoietic stem cell genome editing. FEBS Lett. 2019, 593, 3623–3648.
- Li, C.; Lieber, A. Adenovirus vectors in hematopoietic stem cell genome editing. FEBS Lett. 2019, 593, 3623-3648.Tasca, F.; Wang, Q.; Goncalves, M. Adenoviral Vectors Meet Gene Editing: Rising A Partnership for the Genomic Engineering of Human Stem Cells and Their Progeny. Cells 2020, 9, 953.
- Tasca, F.; Wang, Q.; Goncalves, M. Adenoviral Vectors Meet Gene Editing: Rising A Partnership for the Genomic Engineering of Human Stem Cells and Their Progeny. Cells 2020, 9, 953.Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nature 2016, 539, 479.
- Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nature 2016, 539, 479.Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117.
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117.Fernandes, L.G.V.; Guaman, L.P.; Vasconcellos, S.A.; Heinemann, M.B.; Picardeau, M.; Nascimento, A.L.T.O. Gene silencing based on RNA-guided catalytically inactive Cas9 (dCas9): A new tool for genetic engineering in Leptospira. Sci. Rep. 2019, 9, 1839.
- Fernandes, L.G.V.; Guaman, L.P.; Vasconcellos, S.A.; Heinemann, M.B.; Picardeau, M.; Nascimento, A.L.T.O. Gene silencing based on RNA-guided catalytically inactive Cas9 (dCas9): A new tool for genetic engineering in Leptospira. Sci. Rep. 2019, 9, 1839.Matharu, N.; Rattanasopha, S.; Tamura, S.; Maliskova, L.; Wang, Y.; Bernard, A.; Hardin, A.; Eckalbar, W.L.; Vaisse, C.; Ahituv, N. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science 2019, 363.
- Matharu, N.; Rattanasopha, S.; Tamura, S.; Maliskova, L.; Wang, Y.; Bernard, A.; Hardin, A.; Eckalbar, W.L.; Vaisse, C.; Ahituv, N. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science 2019, 363, doi:10.1126/science.aau0629.Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control Release 2017, 266, 17–26.
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control Release 2017, 266, 17–26.Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424.
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424.Baker, A.T.; Aguirre-Hernandez, C.; Hallden, G.; Parker, A.L. Designer Oncolytic Adenovirus: Coming of Age. Cancers 2018, 10(6).
- Michihiko, S.; Yamamoto, N.; Shinozaki, F.; Shimada, K.; Soma, G.I.; Kobayashi, N. Augmentation of in-vitro HIV replication in peripheral blood mononuclear cells of AIDS and ARC patients by tumour necrosis factor. Lancet 1989, 1, 1206–1207.Fujiwara, T.; Shirakawa, Y.; Kagawa, S. Telomerase-Specific Oncolytic Virotherapy for Human Gastrointestinal Cancer. Expert Rev. Anticancer Ther. 2011, 11, 525–532.
- Fujiwara, T.; Shirakawa, Y.; Kagawa, S. Telomerase-Specific Oncolytic Virotherapy for Human Gastrointestinal Cancer. Expert Rev. Anticancer Ther. 2011, 11, 525–532.Fujiwara, T. A novel molecular therapy using bioengineered adenovirus for human gastrointestinal cancer. Acta Med. Okayama 2011, 65, 151–162.
- Fujiwara, T. A novel molecular therapy using bioengineered adenovirus for human gastrointestinal cancer. Acta Med. Okayama 2011, 65, 151–162.Larson, C.; Oronsky, B.; Scicinski, J.; Fanger, G.R.; Stirn, M.; Oronsky, A.; Reid, T.R. Going viral: A review of replication-selective oncolytic adenoviruses. Oncotarget 2015, 6, 19976–19989.
- Larson, C.; Oronsky, B.; Scicinski, J.; Fanger, G.R.; Stirn, M.; Oronsky, A.; Reid, T.R. Going viral: A review of replication-selective oncolytic adenoviruses. Oncotarget 2015, 6, 19976–19989.Howe, J.A.; Demers, G.W.; Johnson, D.E.; Neugebauer, S.E.; Perry, S.T.; Vaillancourt, M.T.; Faha, B. Evaluation of E1-mutant adenoviruses as conditionally replicating agents for cancer therapy. Mol. Ther. 2000, 2, 485–495.
- Howe, J.A.; Demers, G.W.; Johnson, D.E.; Neugebauer, S.E.; Perry, S.T.; Vaillancourt, M.T.; Faha, B. Evaluation of E1-mutant adenoviruses as conditionally replicating agents for cancer therapy. Mol. Ther. 2000, 2, 485–495.White, E. Regulation of Apoptosis by Adenovirus E1A and E1B Oncogenes. Semin. Virol. 1998, 8, 505–513.
- White, E. Regulation of Apoptosis by Adenovirus E1A and E1B Oncogenes. Semin. Virol. 1998, 8, 505–513.Marcellus, R.C.; Teodoro, J.G.; Charbonneau, R.; Shore, G.C.; Branton, P.E. Expression of p53 in Saos-2 osteosarcoma cells induces apoptosis which can be inhibited by Bcl-2 or the adenovirus E1B-55 kDa protein. Cell Growth Differ. 1996, 7, 1643–1650.
- Marcellus, R.C.; Teodoro, J.G.; Charbonneau, R.; Shore, G.C.; Branton, P.E. Expression of p53 in Saos-2 osteosarcoma cells induces apoptosis which can be inhibited by Bcl-2 or the adenovirus E1B-55 kDa protein. Cell Growth Differ. 1996, 7, 1643–1650.Harada, J.N.; Berk, A.J. p53-Independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J. Virol. 1999, 73, 5333–5344.
- Harada, J.N.; Berk, A.J. p53-Independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J. Virol. 1999, 73, 5333–5344.Wang, H.G.; Draetta, G.; Moran, E. E1A induces phosphorylation of the retinoblastoma protein independently of direct physical association between the E1A and retinoblastoma products. Mol. Cell. Biol. 1991, 11, 4253–4265.
- Wang, H.G.; Draetta, G.; Moran, E. E1A induces phosphorylation of the retinoblastoma protein independently of direct physical association between the E1A and retinoblastoma products. Mol. Cell. Biol. 1991, 11, 4253–4265.Howe, J.A.; Bayley, S.T. Effects of Ad5 E1A mutant viruses on the cell cycle in relation to the binding of cellular proteins including the retinoblastoma protein and cyclin A. Virology 1992, 186, 15–24.
- Howe, J.A.; Bayley, S.T. Effects of Ad5 E1A mutant viruses on the cell cycle in relation to the binding of cellular proteins including the retinoblastoma protein and cyclin A. Virology 1992, 186, 15-24.Eager, R.M.; Nemunaitis, J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 2011, 18, 305–317.
- Eager, R.M.; Nemunaitis, J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 2011, 18, 305–317.Liu, C.-C.; Liu, J.H.; Wu, S.C.; Yen, C.C.; Chen, W.S.; Tsai, Y.C. A Novel E1B-55kD-Deleted Oncolytic Adenovirus Carrying Mutant KRAS-Regulated hdm2 Transgene Exerts Specific Antitumor Efficacy on Colorectal Cancer Cells. Mol. Cancer Ther. 2010, 9, 450–460.
- Liu, C.-C.; Liu, J.H.; Wu, S.C.; Yen, C.C.; Chen, W.S.; Tsai, Y.C. A Novel E1B-55kD-Deleted Oncolytic Adenovirus Carrying Mutant KRAS-Regulated hdm2 Transgene Exerts Specific Antitumor Efficacy on Colorectal Cancer Cells. Mol. Cancer Ther. 2010, 9, 450–460.Kim, J.; Kim, P.-H.; Yoo, J.Y.; Yoon, A.-R.; Choi, H.J.; Seong, J.; Kim, I.-W.; Kim, J.-H.; Yun, C.-O. Double E1B 19 kDa- and E1B 55 kDa-deleted oncolytic adenovirus in combination with radiotherapy elicits an enhanced anti-tumor effect. Gene Ther. 2009, 16, 1111–1121.
- Kim, J.; P.-H.; Kim, Yoo, J.Y.; Yoon, A.-R.; Choi, H.J.; Seong, J.; Kim, I.-W.; Kim, J.-H.; Yun, C.-O. Double E1B 19 kDa- and E1B 55 kDa-deleted oncolytic adenovirus in combination with radiotherapy elicits an enhanced anti-tumor effect. Gene Ther. 2009, 16, 1111–1121.Markedly Enhanced Cytolysis by E1B-19kD-Deleted Oncolytic Adenovirus in Combination with Cisplatin. Hum. Gene Ther. 2006, 17, 379–390.
- Markedly Enhanced Cytolysis by E1B-19kD-Deleted Oncolytic Adenovirus in Combination with Cisplatin. Hum. Gene Ther. 2006, 17, 379–390.Tagliaferri, P.; Caraglia, M.; Budillon, A.; Marra, M.; Vitale, G.; Viscomi, C.; Masciari, S.; Tassone, P.; Abbruzzese, A.; Venuta, S. New pharmacokinetic and pharmacodynamic tools for interferon-alpha (IFN-alpha) treatment of human cancer. Cancer Immunol. Immunother. 2005, 54, 1–10.
- Tagliaferri, P.; Caraglia, M.; Budillon, A.; Marra, M.; Vitale, G.; Viscomi, C.; Masciari, S.; Tassone, P.; Abbruzzese, A.; Venuta, S. New pharmacokinetic and pharmacodynamic tools for interferon-alpha (IFN-alpha) treatment of human cancer. Cancer Immunol. Immunother. 2005, 54, 1–10.Nelson, A.R.; Davydova, J.; Curiel, D.T.; Yamamoto, M. Combination of conditionally replicative adenovirus and standard chemotherapies shows synergistic antitumor effect in pancreatic cancer. Cancer Sci. 2009, 100, 2181–2187.
- Nelson, A.R.; Davydova, J.; Curiel, D.T.; Yamamoto, M. Combination of conditionally replicative adenovirus and standard chemotherapies shows synergistic antitumor effect in pancreatic cancer. Cancer Sci. 2009, 100, 2181–7.Lakdawala, S.S.; Schwartz, R.A.; Ferenchak, K.; Carson, C.T.; McSharry, B.P.; Wilkinson, G.W.; Weitzman, M.D. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J. Virol. 2008, 82, 8362–8372.
- Lakdawala, S.S.; Schwartz, R.A.; Ferenchak, K.; Carson, C.T.; McSharry, B.P.; Wilkinson, G.W.; Weitzman, M.D. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J. Virol. 2008, 82, 8362–8372.Carson, C.T.; Orazio, N.I.; Lee, D.V.; Suh, J.; Bekker-Jensen, S.; Araujo, F.D.; Lakdawala, S.S.; Lilley, C.E.; Bartek, J.; Lukas, J. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009, 28, 652–662.
- Carson, C.T.; Orazio, N.I.; Lee, D.V.; Suh, J.; Bekker-Jensen, S.; Araujo, F.D.; Lakdawala, S.S.; Lilley, C.E.; Bartek, J.; Lukas, J. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009, 28, 652–662.Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650.
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650.
- Rizvi, S.A.A.; Shahzad, Y.; Saleh, A.M.; Muhammad, N. Dose Issues in Cancer Chemotherapy. Oncology 2020, 1–8, doi:10.1159/000506705.
- Haley, B.; Roudnicky, F. Functional Genomics for Cancer Drug Target Discovery. Cancer Cell 2020, doi:10.1016/j.ccell.2020.04.006.
- Roy, A. Early Probe and Drug Discovery in Academia: A Minireview. High Throughput 2018, 7, 4.
- Von Stechow, L.; Cancer Systems Biology: Methods and Protocols; Springer: New York, NY, USA, 2018.
- Kibble, M.; Saarinen, N.; Tang, J.; Wennerberg, K.; Mäkelä, S.; Aittokallio, T. Network pharmacology applications to map the unexplored target space and therapeutic potential of natural products. Nat. Prod. Rep. 2015, 32, 1249–66.
- Barneh, F.; Jafari, M.; Mirzaie, M. Updates on drug-target network; facilitating polypharmacology and data integration by growth of DrugBank database. Brief Bioinform.2016, 17, 1070–1080.
- Smith, R.D.; Hu, L.; Falkner, J.A.; Benson, M.L.; Nerothin, J.P.; Carlson, H.A. Exploring protein-ligand recognition with Binding MOAD. J. Mol. Graph. Model 2006, 24, 414–425, doi:10.1016/j.jmgm.2005.08.002.
- Hermiston, T.W.; Kuhn, I. Armed therapeutic viruses: Strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002,9 1022-35.
- Davydova, J.; Yamamoto, M. Oncolytic adenoviruses: Design, generation, and experimental procedures. Curr. Protoc. Hum. Genet. 2013, 78, 12.14.1–12.14.2.
- Lauer, U.M.; Schell, M.; Beil, J.; Berchtold, S.; Koppenhöfer, U.; Glatzle, J.; Königsrainer, A.; Möhle, R.; Nann, D.; Fend, F. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clin. Cancer Res.2018, 24, 4388–4398.
- Kemp, V.; Hoeben, R.C.; van den Wollenberg, D.J. Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses 2015, 8, 4, doi:10.3390/v8010004.
- Zhao, X.; Cariad Chester, Rajasekaran, N.; He, Z.X.; Kohrt, H.E. Strategic Combinations: The Future of Oncolytic Virotherapy with Reovirus. Mol. Cancer 2016, 15, 767–773.
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, Gu.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158.
- Stone, D.; Liu, Y.; Shayakhmetov, D.; Li, Zo.; Ni, S.; Lieber, A. Adenovirus-platelet interaction in blood causes virus sequestration to the reticuloendothelial system of the liver. J. Virol. 2007, 81, 4866–4871.
- Shayakhmetov, D.M.; Eberly, A.M.; Li, Zo.; Lieber, A. Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J. Virol. 2005, 79, 1053–1061.
- Meckes, D.G.; Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol.2011, 85, 12844–12854.
- Yao, X.L.; Yoshioka, Y.; Ruan, G.; Chen, Y.; Mizuguchi, H.; Mukai, Y.; Okada, N.; Gao, Ji.; Nakagawa, S. Optimization and internalization mechanisms of PEGylated adenovirus vector with targeting peptide for cancer gene therapy. Biomacromolecules 2012, 13, 2402–2409.
- Ghosh, D.; Barry, M.A. Selection of muscle-binding peptides from context-specific peptide-presenting phage libraries for adenoviral vector targeting. J. Virol.2005, 79, 13667–13672.
- Shashkova, E.V.; May, S.M.; Doronin, K.; Barry. M.A. Expanded anticancer therapeutic window of hexon-modified oncolytic adenovirus. Mol. Ther. 2009, 17, 2121–2130.
- Chen, Z.; Mok, H.; Pflugfelder, S.C.; Li, D.Q.; Barry, M.A. Improved transduction of human corneal epithelial progenitor cells with cell-targeting adenoviral vectors. Experimental eye research 2006, 83, 798–806, doi:10.1016/j.exer.2006.03.023.
- Korokhov, N.; Mikheeva, G.; Krendelshchikov, A.; Belousova, N.; Simonenko, V.; Krendelshchikova, V.; Pereboev, A.; Kotov, A.; Kotova, O.; Triozzi, P.L., et al. Targeting of adenovirus via genetic modification of the viral capsid combined with a protein bridge. J Virol 2003, 77, 12931–12940, doi:10.1128/jvi.77.24.12931-12940.2003.
- Volpers, C.; Thirion, C.; Biermann, V.; Hussmann, S.; Kewes, H.; Dunant, P.; von der Mark, H.; Herrmann, A.; Kochanek, S.; Lochmuller, H. Antibody-mediated targeting of an adenovirus vector modified to contain a synthetic immunoglobulin g-binding domain in the capsid. J Virol 2003, 77, 2093–2104, doi:10.1128/jvi.77.3.2093-2104.2003.
- Wickham, T.J.; Lee, G.M.; Titus, J.A.; Sconocchia, G.; Bakacs, T.; Kovesdi, I.; Segal, D.M. Targeted adenovirus-mediated gene delivery to T cells via CD3. J Virol 1997, 71, 7663–7669.
- Borovjagin, A.V.; Krendelchtchikov, A.; Ramesh, N.; Yu, D.C.; Douglas, J.T.; Curiel, D.T. Complex mosaicism is a novel approach to infectivity enhancement of adenovirus type 5-based vectors. Cancer Gene Ther. 2005, 12, 475–486, doi:10.1038/sj.cgt.7700806.
- Sakurai, F. Development and evaluation of a novel gene delivery vehicle composed of adenovirus serotype 35. Biol. Pharm. Bull. 2008, 31, 1819–1825, doi:10.1248/bpb.31.1819.
- Preuss, M.A.; Glasgow, J.N.; Everts, M.; Stoff-Khalili, M.A.; Wu, H.; Curiel, D.T. Enhanced Gene Delivery to Human Primary Endothelial Cells Using Tropism-Modified Adenovirus Vectors. Open Gene Ther. J. 2008, 1, 7–11, doi:10.2174/1875037000801010007.
- Yang, M.; Yang, C.S.; Guo, W.; Tang, J.; Huang, Q.; Feng, S.; Jiang, A.; Xu, X.; Jiang, G.; Liu, Y.Q. A novel fiber chimeric conditionally replicative adenovirus-Ad5/F35 for tumor therapy. Cancer Biol. Ther. 2017, 18, 833–840, doi:10.1080/15384047.2017.1395115.
- Kanerva, A.; Mikheeva, G.V.; Krasnykh, V.; Coolidge, C.J.; Lam, J.T.; Mahasreshti, P.J.; Barker, S.D.; Straughn, M.; Barnes, M.N.; Alvarez, R.D., et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin Cancer Res 2002, 8, 275–280.
- Guse, K.; Ranki, T.; Ala-Opas, M.; Bono, P.; Sarkioja, M.; Rajecki, M.; Kanerva, A.; Hakkarainen, T.; Hemminki, A. Treatment of metastatic renal cancer with capsid-modified oncolytic adenoviruses. Mol. Cancer The. 2007, 6, 2728–2736, doi:10.1158/1535-7163.MCT-07-0176.
- Wei, X.; Liu, L.; Wang, G.; Li, W.; Xu, K.; Qi, H.; Liu, H.; Shen, J.; Li, Z.; Shao, J. Potent antitumor activity of the Ad5/11 chimeric oncolytic adenovirus combined with interleukin-24 for acute myeloid leukemia via induction of apoptosis. Oncol. Rep. 2015, 33, 111–118, doi:10.3892/or.2014.3563.
- Machiels, J.P.; Salazar, R.; Rottey, S.; Duran, I.; Dirix, L.; Geboes, K.; Wilkinson-Blanc, C.; Pover, G.; Alvis, S.; Champion, B., et al. A phase 1 dose escalation study of the oncolytic adenovirus enadenotucirev, administered intravenously to patients with epithelial solid tumors (EVOLVE). J. Immunother. Cancer 2019, 7, 20, doi:10.1186/s40425-019-0510-7.
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K., et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther 2010, 18, 1874–1884, doi:10.1038/mt.2010.161.
- Kim, K.H.; Dmitriev, I.P.; Saddekni, S.; Kashentseva, E.A.; Harris, R.D.; Aurigemma, R.; Bae, S.; Singh, K.P.; Siegal, G.P.; Curiel, D.T., et al. A phase I clinical trial of Ad5/3-Delta24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol. Oncol. 2013, 130, 518–524., doi:10.1016/j.ygyno.2013.06.003.
- Seki, T.; Dmitriev, I.; Suzuki, K.; Kashentseva, E.; Takayama, K.; Rots, M.; Uil, T.; Wu, H.; Wang, M.; Curiel, D.T. Fiber shaft extension in combination with HI loop ligands augments infectivity for CAR-negative tumor targets but does not enhance hepatotropism in vivo. Gene Ther. 2002, 9, 1101–1108., doi:10.1038/sj.gt.3301815.
- Janzen, W. High Throughput Screening: Methods and Protocols; Humana Press: New York, NY, USA, 2016; Volume 1439.
- Hust, M.; Lim, T.S. Phage display: Methods and Protocols; Humana Press: New York, NY, USA, 2018.
- Bhatia, S.; O'Bryan, S.M.; Rivera, A.A.; Curiel, D.T.; Mathis, J.M. CXCL12 retargeting of an adenovirus vector to cancer cells using a bispecific adapter. Oncolytic Virother. 2016, 5, 99–113.
- Kloos, A.; Woller, N.; Gürlevik, E.; Ureche, Cr.; Niemann, J.; Armbrecht, N.; Martin, N.T.; Geffers, R.; Manns, M.P.; Gerardy-Schahn, R. PolySia-Specific Retargeting of Oncolytic Viruses Triggers Tumor-Specific Immune Responses and Facilitates Therapy of Disseminated Lung Cancer. Cancer Immunol. Res. 2015, 3, 751–763.
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; jan Krönke; Facon, T.; Salnikov, A.V.; Lesley, R.; Beutner, K. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783.
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Jr.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616.
- Bramante, S.; Kaufmann, J.K.; Veckman, V.; Liikanen, I.; Nettelbeck, D.M.; Hemminki, O.; Vassilev, L.; Cerullo, V.; Oksanen, M.; Heiskanen, R., et al. Treatment of melanoma with a serotype 5/3 chimeric oncolytic adenovirus coding for GM-CSF: Results in vitro, in rodents and in humans. International journal of cancer 2015, 137, 1775-1783, doi:10.1002/ijc.29536.
- Flemington, E.K.; Speck, S.H.; Kaelin, W.G.; Jr. E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc. Natl. Acad. Sci. USA 1993, 90, 6914–6918.
- Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J. Urol. 2012, 188, 2391–2397.
- Boettcher, M. and M.T. McManus, Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585.
- Hajeri, P.B.; Singh, S.K. Rna Interference: From basics to Therapeutics. In Molecular and Cellular Therapeutics; Whitehouse, D., Rapley, R., Eds.; Wiley: Hoboken, NJ, USA, 2012; pp. 140–167.
- Hajeri, P.B.; Singh, S.K. siRNAs: Their potential as therapeutic agents--Part, I. Designing of siRNAs. Drug Discov. Today 2009, 14, 851–858.
- Singh, S.K.; Hajeri, P.B. siRNAs: Their potential as therapeutic agents--Part II. Methods of delivery. Drug Discov. Today 2009, 14, 859–865.
- Liu, Y.P.; Berkhout, B.; miRNA cassettes in viral vectors: Problems and solutions. Biochim Biophys Acta 2011, 1809, 732–745.
- Callegari, E.; Elamin, B.K.; D'Abundo, L.; Falzoni, S.; Donvito, G.; Moshiri, F.; Milazzo, M.; Altavilla, G.; Giacomelli, L.; Fornari, F. Anti-tumor activity of a miR-199-dependent oncolytic adenovirus. PLoS ONE 2013, 8, e73964.
- Lou, W.; Chen, Q.; Ma, L.; Liu, J.; Yang, Z.; Shen, J.; Cui, Y.; Bian, Xi.; Qian, C. Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. J. Mol. Med. 2013, 91, 715–725.
- Kosaka, T.; Davydova, J.; Ono, H.A.; Akiyama, H.; Hirai, Sh.; Ohno, S.; Takeshita, F.; Aoki, K.; Ochiya, T.; Yamamoto, M.; Kunisaki, C. Imaging and Antitumoral Effect of a Cyclo-oxygenase 2-specific Replicative Adenovirus for Small Metastatic Gastric Cancer Lesions. Anticancer Res. 2015, 35, 5201–5210.
- Cui, C.X.; Li, Y.-Q.; Sun, Y.-J.; Zhu, Y.-L.; Fang, J.-B.; Bai, B.; Li, W.-J.; Li, S.-Z.; Ma, Y.-Z.; Li, X. Antitumor effect of a dual cancer-specific oncolytic adenovirus on prostate cancer PC-3 cells. Urol. Oncol. 2019, 37, 352 e1–352 e18.
- Lu, S.; Cullen, B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004, 78, 12868–12876.
- Yang, Z.; Li, Y. Dissection of RNAi-based antiviral immunity in plants. Curr. Opin. Virol. 2018, 32, 88–99.
- Schuster, S.; P. Miesen; van Rij, R.P. Antiviral RNAi in Insects and Mammals: Parallels and Differences. Viruses 2019, 11, 448.
- Zhao, J.H.; Hua, C.L.; Fang, Y.Y.; Guo, H.S. The dual edge of RNA silencing suppressors in the virus-host interactions. Curr. Opin. Virol. 2016, 17, 39–44.
- Machitani, M.; Sakurai, F.; Katayama, K.; Tachibana, M.; Suzuki, T.; Matsui, H.; Yamaguchi, T.; Mizuguchi, H. Improving adenovirus vector-mediated RNAi efficiency by lacking the expression of virus-associated RNAs. Virus Res. 2013, 178, 357–363.
- Aparicio, O.; et al. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J. Virol. 2006, 80, 1376–1384.
- Bennasser, Y.; Chable-Bessia, C.; Triboulet, R.; Gibbings, D.; Gwizdek, C.; Dargemont, C.; Kremer, E.J.; Voinnet, O.; Benkirane, M. Competition for XPO5 binding between Dicer mRNA, pre-miRNA and viral RNA regulates human Dicer levels. Nat. Struct. Mol. Biol. 2011, 18, 323–327.
- Machitani, M.; Yamaguchi, T.; Shimizu, K.; Sakurai, F.; Katayama, K.; Kawabata, K.; Mizuguchi, H. Adenovirus Vector-Derived VA-RNA-Mediated Innate Immune Responses. Pharmaceutics 2011, 3, 338–353.
- Kitajewski, J.; Schneider, R.J.; Safer, B.; Munemitsu, S.M.; Samuel, C.E.; Thimmappaya, B.; Shenk, T. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 1986, 45, 195–200.
- Vachon, V.K.; Conn, G.L. Adenovirus VA RNA: An essential pro-viral non-coding RNA. Virus Res. 2016, 212, 39–52.
- Carette, J.E.; Overmeer, R.M.; Schagen, F.H.; Alemany, R.; Barski, O.A.; Gerritsen, W.R.; Van Beusechem, V.W. Conditionally replicating adenoviruses expressing short hairpin RNAs silence the expression of a target gene in cancer cells. Cancer Res. 2004, 64, 2663–2667.
- Rovira-Rigau, M.; Raimondi, G.; Marín, M.Á.; Gironella, M.; Alemany, R.; Fillat, C. Bioselection Reveals miR-99b and miR-485 as Enhancers of Adenoviral Oncolysis in Pancreatic Cancer. Mol. Ther. 2019, 27, 230–243.
- Brachtlova, T.; van Beusechem, V.W. Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens. Cells 2018, 7, 288.
- Jin, H.; Lv, S.; Yang, J.; Wang, X.; Hu, H.; Su, C.; Zhou, C.; Li, J.; Huang, Y.; Li, L. Use of microRNA Let-7 to control the replication specificity of oncolytic adenovirus in hepatocellular carcinoma cells. PLoS ONE 2011, 6, e21307.
- Karimian, A.; Azizian, K.; Parsian, H.; Rafieian, S.; Shafiei-Irannejad, V.; Kheyrollah, M.; Yousefi, M.; Majidinia, M.; Yousefi, B. CRISPR/Cas9 technology as a potent molecular tool for gene therapy. J. Cell Physiol. 2019, 234, 12267-12277.
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct Target Ther. 2020, 5, 1.
- Li, C.; Lieber, A. Adenovirus vectors in hematopoietic stem cell genome editing. FEBS Lett. 2019, 593, 3623-3648.
- Matharu, N.; Rattanasopha, S.; Tamura, S.; Maliskova, L.; Wang, Y.; Bernard, A.; Hardin, A.; Eckalbar, W.L.; Vaisse, C.; Ahituv, N. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science 2019, 363, doi:10.1126/science.aau0629.
- Michihiko, S.; Yamamoto, N.; Shinozaki, F.; Shimada, K.; Soma, G.I.; Kobayashi, N. Augmentation of in-vitro HIV replication in peripheral blood mononuclear cells of AIDS and ARC patients by tumour necrosis factor. Lancet 1989, 1, 1206–1207.
- Howe, J.A.; Bayley, S.T. Effects of Ad5 E1A mutant viruses on the cell cycle in relation to the binding of cellular proteins including the retinoblastoma protein and cyclin A. Virology 1992, 186, 15-24.
- Kim, J.; P.-H.; Kim, Yoo, J.Y.; Yoon, A.-R.; Choi, H.J.; Seong, J.; Kim, I.-W.; Kim, J.-H.; Yun, C.-O. Double E1B 19 kDa- and E1B 55 kDa-deleted oncolytic adenovirus in combination with radiotherapy elicits an enhanced anti-tumor effect. Gene Ther. 2009, 16, 1111–1121.
- Nelson, A.R.; Davydova, J.; Curiel, D.T.; Yamamoto, M. Combination of conditionally replicative adenovirus and standard chemotherapies shows synergistic antitumor effect in pancreatic cancer. Cancer Sci. 2009, 100, 2181–7.