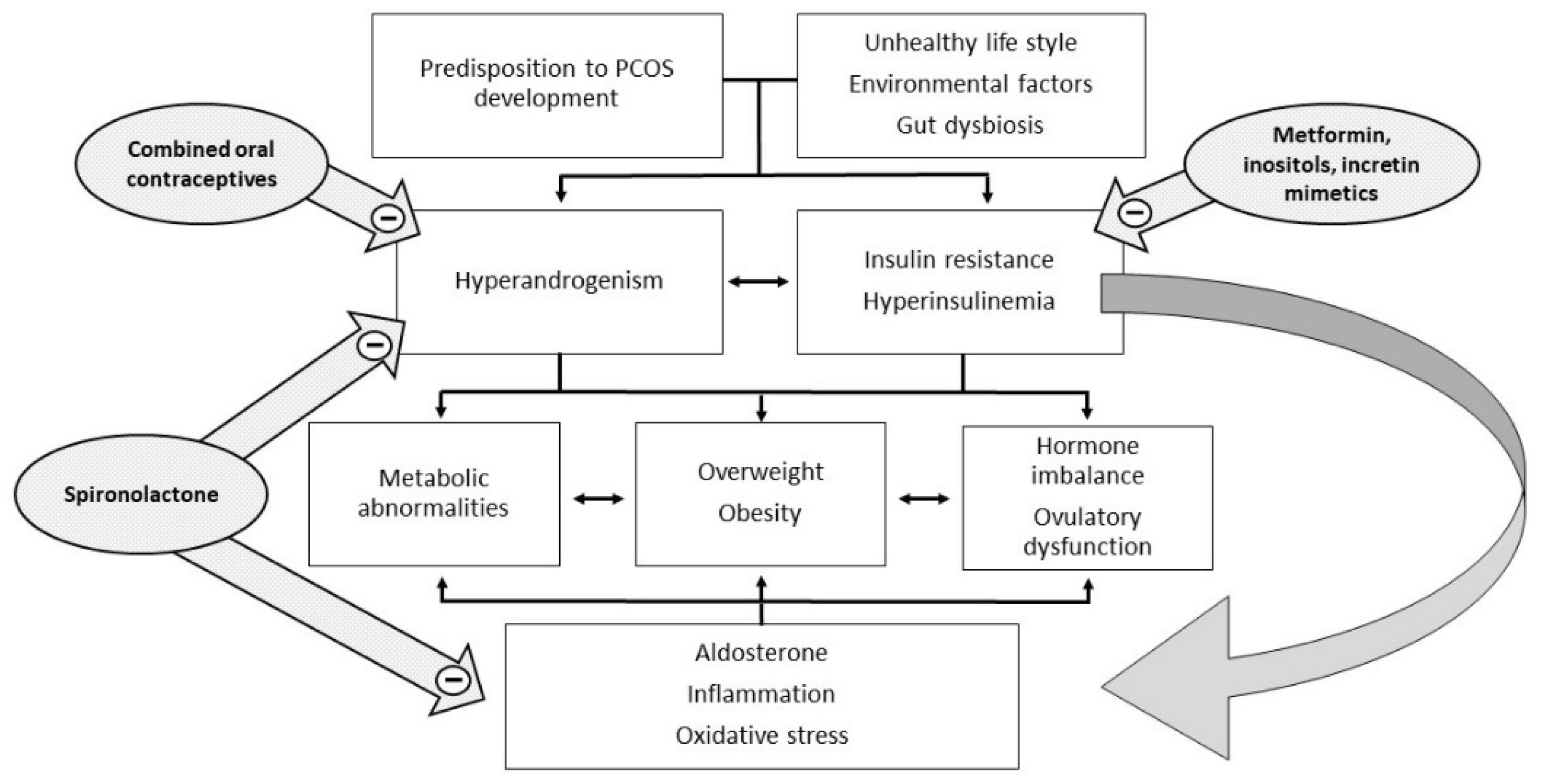
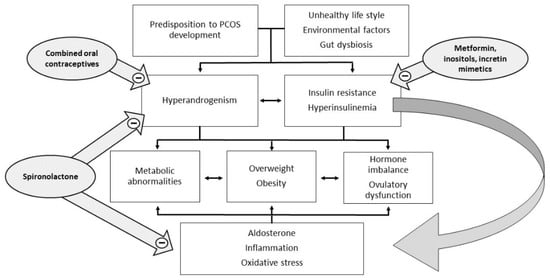
Polycystic ovary syndrome (PCOS) is a heterogeneous and extremely common disease with symptoms that vary with the age of the patient, typically characterized by hyperandrogenism, chronic oligo-anovulation, and/or several metabolic disorders. The syndrome includes various phenotypes, and the pathogenesis is multifactorial, often involving insulin resistance. This feature is closely related to ovarian dysfunction, inflammation, hyperandrogenism, and metabolic disorders, which characterize and complicate the syndrome. Therapy currently considers both lifestyle improvements and medications, and must be tailored on a case-by-case basis. To date, the published studies have not arrived at a definition of the most suitable therapy for each individual case and many of the drugs used are still off-label.
- polycystic ovary syndrome
- insulin resistance
- aldosterone
- hyperandrogenism
- metformin
- inositol
- insulin sensitizers
- spironolactone
1. Introduction
2. The Pathogenetic Role of Insulin Resistance in PCOS
Insulin acts as a regulator of glucose homeostasis by stimulating glucose uptake by insulin-sensitive tissues, such as adipose tissue, skeletal muscle, liver, and heart, but also by suppressing hepatic glucose production. Insulin is also able to suppress lipolysis, leading to a decrease in free fatty acid levels, which may mediate insulin’s action on hepatic glucose production. Insulin resistance is defined as a decreased ability of insulin to carry out these metabolic actions inherent in glucose uptake and production and lipolysis, thus leading to compensatory high insulin levels, both at baseline and after glucose loading, if pancreatic function is normal. There is still no consensus on the exact mechanism that leads to insulin resistance in PCOS, regardless of body mass index (BMI). An old study argued that in PCOS, the mechanism underlying insulin resistance decreased autophosphorylation of the insulin receptor following insulin binding [5]. The mechanisms by which insulin resistance exerts its effects have only recently been well described [6]. At a liver and skeletal muscle level, insulin resistance increases lipolysis with the accumulation of non-esterified fatty acids. The accumulation of intrahepatic lipids activates the diacylglycerol/protein kinase C axis and inhibits the insulin receptor, also affecting insulin signaling and subsequent gluconeogenesis. In skeletal muscle, the inhibition of phosphoinositide-3 kinase and phosphorylation of insulin receptor substrate 1 leads to impaired insulin signaling by altering the GLUT-4 expression and glucose uptake [6][7][6,7]. The consequence of hyperinsulinemia compensatory to insulin resistance is an overstimulation of non-insulin-sensitive tissues, such as the ovaries. In particular, insulin and LH act synergistically on the theca cells, stimulating ovarian androgen production [4]. In addition, insulin acts both directly as a co-gonadotropin, enhancing LH activity by stimulating the expression of receptors for LH, insulin, and IGF on granulosa cells, and indirectly by impairing the regulation of the hypothalamic–pituitary–ovarian axis. Hyperinsulinemia increases the adrenal steroid response to ACTH stimuli and decreases the synthesis of sex hormone binding globulin (SHBG) in the liver, with a consequent increase of both total and free androgen levels (Figure 1).