The most of the transportations in cells are realized through a kind of proteins, the molecular motor. Molecular motor can be classed into three families, myosin, kinesin and dynein. Kinesin-1 (also called conventional kinesin) is the founding member of the kinesin family and mainly exists in the nerve axons to transport membranous organelles along the microtubule lattice. By using the energy stored in the ATP molecule, kinesin-1 can “walk” along the microtubule lattice in a hand-over-hand manner. In the walking process of the kinesin-1, the conformational changes of the compact motor domain transmit and amplify the small changes of the nucleotide-binding site to the force-generation element to produce the processive movement. The chemical cycle and mechanical cycle of kinesin-1 are highly coupled to ensure the processivity of the kinesin-1 and to avoid the futile ATP hydrolysis.
- Kinesin-1, neck linker, nucleotide
1. Introduction
Kinesin is a molecular walking motor protein inside cells. The size of kinesin is smaller than the other two members of motor proteins, myosin and dynein. The kinesin proteins can be divided into 14 subfamilies according to their structural and functional similarities (from kinesin-1 to kinesin-14) [1][2][3][4]. The kinesin-1 subfamily (also called conventional kinesin) is the founding member of the kinesin family [5][6] and mainly exists in the nerve axons to transport membranous organelles along microtubule lattice. Different from the kinesin-3 (monomer, but can also form a dimer [7]) and the kinesin-5 (tetramer) subfamily, the members of kinesin-1 form a dimer structure in vivo to “walk” toward the microtubule’s plus end. The entire structure of kinesin-1 can be mainly divided into three domains, i.e., the motor domain, the tail domain and the stalk domain (the motor domain and a part of stalk domain of kinesin-1 are shown in
Kinesin is a molecular walking motor protein inside cells. The size of kinesin is smaller than the other two members of motor proteins, myosin and dynein. The kinesin proteins can be divided into 14 subfamilies according to their structural and functional similarities (from kinesin-1 to kinesin-14) [1,2,3,4]. The kinesin-1 subfamily (also called conventional kinesin) is the founding member of the kinesin family [5,6] and mainly exists in the nerve axons to transport membranous organelles along microtubule lattice. Different from the kinesin-3 (monomer, but can also form a dimer [7]) and the kinesin-5 (tetramer) subfamily, the members of kinesin-1 form a dimer structure in vivo to “walk” toward the microtubule’s plus end. The entire structure of kinesin-1 can be mainly divided into three domains, i.e., the motor domain, the tail domain and the stalk domain (the motor domain and a part of stalk domain of kinesin-1 are shown in
Figure 1). The motor domain (also called motor head), which contains the nucleotide-binding and microtubule-binding sites, is highly conserved among kinesin family. The tail domain of kinesin-1 is used to bind with the “cargo”. Kinesin-1 proteins have different tail domains, which can bind the light chain to interact with different cargos [8]. The motor domain and the tail domain are connected by a single long α-helix, which is called the stalk domain. The two stalk domains of two kinesin-1 monomers coil together to form a coiled-coil structure and constitute a functional dimer. It is worthy to note that ~14 residues constitute the neck linker of kinesin-1, which connects the motor domain and the stalk domain. The conformational changes of the neck linker in different nucleotide-binding states are the key processes in the walking movement of kinesin-1 [9][10][11][12][13]. Because the motor domain locates in the N-terminal part of the protein, kinesin-1 belongs to the N-type kinesin. Kinesins with motor domain located in the middle and the C-terminal part of the protein are the M-type and C-type, respectively. The walking directionality of kinesin varies with different locations of the motor domain. N-type kinesins (most members of the kinesin family) walk toward the plus end of the microtubule (Some of the kinesin-5 proteins with N-terminal motor domain show bidirectional motility, as reviewed in Ref. [14]). In contrast, the C-type kinesin (mainly kinesin-14 subfamily [15]) walks toward the minus end of the microtubule. The M-type kinesin (mainly kinesin-13 subfamily) is relatively special because it takes one-dimensional diffusion toward the two ends of the microtubule [16][17][18][19]. The kinesin-1 dimer walks along a single protofilament of the microtubule in a hand-over-hand manner. There are some noteworthy features of kinesin-1 walking movement: 1) Kinesin-1 can transform chemical energy of the adenosine triphosphate (ATP) binding and hydrolysis to mechanical energy of the walking along the microtubule with a cargo. 2) The chemical cycle and the mechanical cycle of kinesin-1 are highly coupled to ensure only one ATP molecule is consumed in one step [20][21]. The futile ATP hydrolysis rarely happens in kinesin-1 normal walking process. 3) The microtubule not only provides the track for the motility of kinesin-1 but also directly participate in the regulation of the kinesin-1 chemical cycle. Microtubule can catalyze the release of the adenosine diphosphate (ADP), which is the product of ATP hydrolysis. In this way, the mechanochemical process of kinesin-1 is dramatically accelerated. The key process of energy transformation is from ATP entering the nucleotide-binding pocket to the docking movement of neck linker, which pulls the other motor domain to the next binding site on the microtubule.
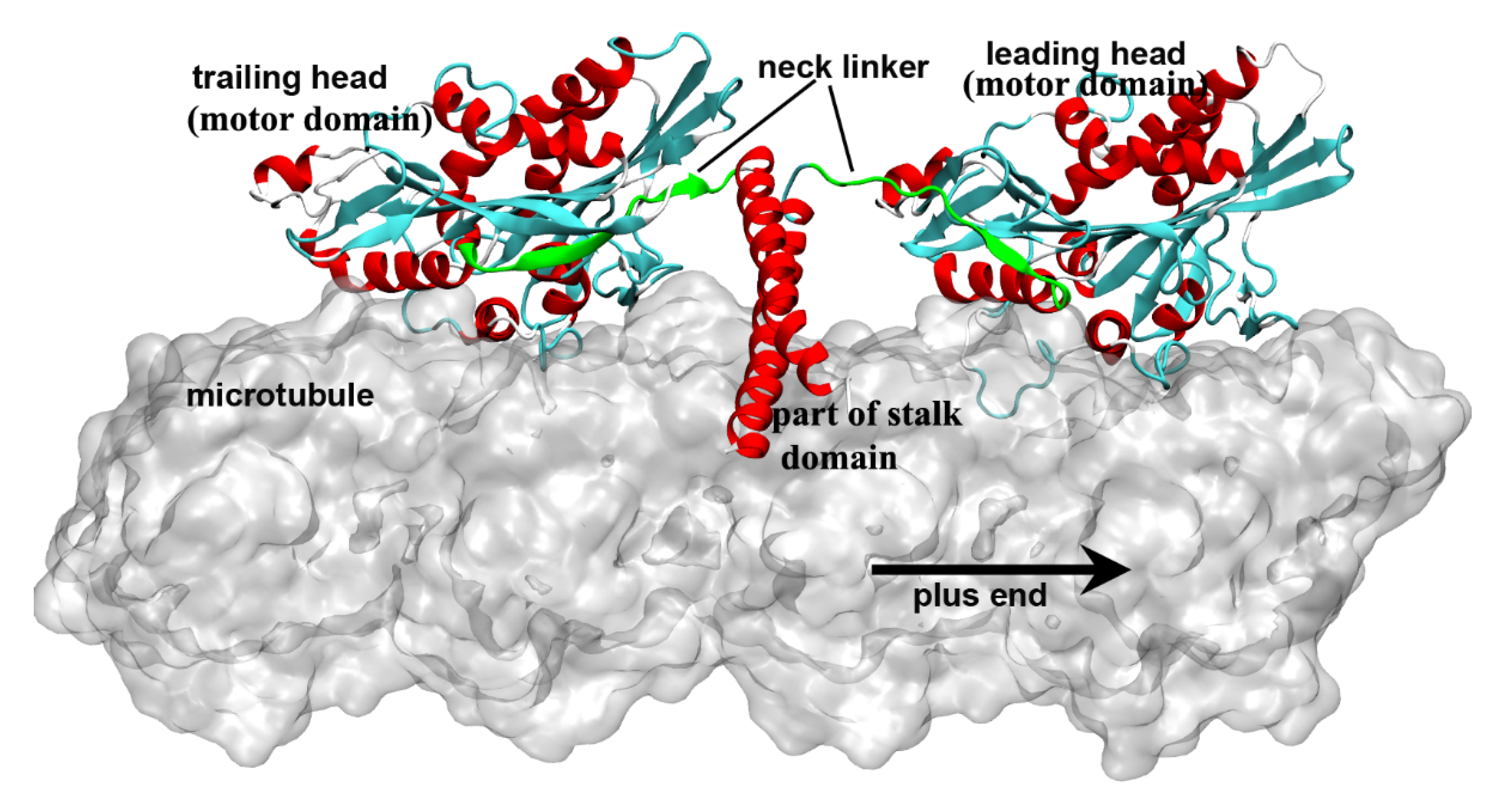
). The motor domain (also called motor head), which contains the nucleotide-binding and microtubule-binding sites, is highly conserved among kinesin family. The tail domain of kinesin-1 is used to bind with the “cargo”. Kinesin-1 proteins have different tail domains, which can bind the light chain to interact with different cargos [8]. The motor domain and the tail domain are connected by a single long α-helix, which is called the stalk domain. The two stalk domains of two kinesin-1 monomers coil together to form a coiled-coil structure and constitute a functional dimer. It is worthy to note that ~14 residues constitute the neck linker of kinesin-1, which connects the motor domain and the stalk domain. The conformational changes of the neck linker in different nucleotide-binding states are the key processes in the walking movement of kinesin-1 [9,10,11,12,13]. Because the motor domain locates in the N-terminal part of the protein, kinesin-1 belongs to the N-type kinesin. Kinesins with motor domain located in the middle and the C-terminal part of the protein are the M-type and C-type, respectively. The walking directionality of kinesin varies with different locations of the motor domain. N-type kinesins (most members of the kinesin family) walk toward the plus end of the microtubule (Some of the kinesin-5 proteins with N-terminal motor domain show bidirectional motility, as reviewed in Ref. [14]). In contrast, the C-type kinesin (mainly kinesin-14 subfamily [15]) walks toward the minus end of the microtubule. The M-type kinesin (mainly kinesin-13 subfamily) is relatively special because it takes one-dimensional diffusion toward the two ends of the microtubule [16,17,18,19]. The kinesin-1 dimer walks along a single protofilament of the microtubule in a hand-over-hand manner. There are some noteworthy features of kinesin-1 walking movement: 1) Kinesin-1 can transform chemical energy of the adenosine triphosphate (ATP) binding and hydrolysis to mechanical energy of the walking along the microtubule with a cargo. 2) The chemical cycle and the mechanical cycle of kinesin-1 are highly coupled to ensure only one ATP molecule is consumed in one step [20, 21]. The futile ATP hydrolysis rarely happens in kinesin-1 normal walking process. 3) The microtubule not only provides the track for the motility of kinesin-1 but also directly participate in the regulation of the kinesin-1 chemical cycle. Microtubule can catalyze the release of the adenosine diphosphate (ADP), which is the product of ATP hydrolysis. In this way, the mechanochemical process of kinesin-1 is dramatically accelerated. The key process of energy transformation is from ATP entering the nucleotide-binding pocket to the docking movement of neck linker, which pulls the other motor domain to the next binding site on the microtubule.
Figure 1. Two motor domains of kinesin-1 bind to the microtubule lattice simultaneously. The neck linkers of the kinesin-1 dimer are colored in green. The leading head is in the nucleotide-free state and has an undocked neck linker, which points to the minus end of the microtubule. The trailing head is in the ADP·Pi/ADP-bound state and has a plus-end pointed neck linker. This figure was produced using Discovery studio 3.5 visualizer.
2. Force-Generation Process of Kinesin-1
Kinesin-1 can catalyze the ATP hydrolysis and use the energy stored in the ATP molecule to realize directional movement [22]. The motor domain of kinesin-1 has a typical globular structure with a central β-sheet of eight strands in the center and three α-helices on either side of the central β-sheet [23][24][25]. The nucleotide-binding and the microtubule-binding sites of kinesin-1 locate on the opposite sides of the central β-sheet. Similar to other ATPase, the nucleotide-binding pocket of kinesin-1 consists of four motifs [26][27], which are referred to as the N-1 motif (also called P-loop or Walker A sequence [28][29], Gly-X-X-X-X-Gly-Lys-Thr/Ser), the N-2 motif (also called switch-Ⅰ, Asn-X-X-Ser-Ser-Arg), the N-3 motif (also called switch-Ⅱ, Asp-X-X-Gly-X-Glu) and the N-4 motif (Arg-X-Arg-Pro). The entire structure of nucleotide-binding pocket of kinesin-1 is a perfect match for ATP molecule in both geometry and interaction. The N-1, N-2 and N-3 motifs form a pocket structure [30]. The three phosphate groups of ATP molecule, which have four negative charges, bind tightly into this pocket and, together with Mg
Kinesin-1 can catalyze the ATP hydrolysis and use the energy stored in the ATP molecule to realize directional movement [22]. The motor domain of kinesin-1 has a typical globular structure with a central β-sheet of eight strands in the center and three α-helices on either side of the central β-sheet [23,24,25]. The nucleotide-binding and the microtubule-binding sites of kinesin-1 locate on the opposite sides of the central β-sheet. Similar to other ATPase, the nucleotide-binding pocket of kinesin-1 consists of four motifs [26, 27], which are referred to as the N-1 motif (also called P-loop or Walker A sequence [28,29], Gly-X-X-X-X-Gly-Lys-Thr/Ser), the N-2 motif (also called switch-Ⅰ, Asn-X-X-Ser-Ser-Arg), the N-3 motif (also called switch-Ⅱ, Asp-X-X-Gly-X-Glu) and the N-4 motif (Arg-X-Arg-Pro). The entire structure of nucleotide-binding pocket of kinesin-1 is a perfect match for ATP molecule in both geometry and interaction. The N-1, N-2 and N-3 motifs form a pocket structure [30]. The three phosphate groups of ATP molecule, which have four negative charges, bind tightly into this pocket and, together with Mg
2+, form salt bridges, hydrogen bonds and coordination bonds with the surrounding residues [30][31][32]. N-4 and N-1 form a geometrically matched hydrophobic pocket for the adenosine ring of ATP. The hydrophobic stacking between the pocket and the adenosine ring of ATP molecule contributes to the binding and recognition of nucleotide with the nucleotide-binding pocket of kinesin-1. The entrance of the ATP molecule into the nucleotide-binding pocket can induce a series of conformational changes of the pocket, which can be amplified and finally transmitted to the neck linker portion to initiate the docking movement of neck linker to the motor domain [12][33][34][35][36][37][38][39][40][41]. The first step of this process is the sensing of the existence of the ATP molecule [42][43]. The switch-Ⅰ and switch-Ⅱ are the γ-phosphate sensors of kinesin-1. Both the motifs have large conformational changes upon ATP binding into the nucleotide-binding pocket of kinesin family [44]. Comparison of structures of motor domain in different nucleotide-binding states shows that the switch-Ⅰ and switch-Ⅱ are in an open conformation in the nucleotide-free state (apo state). After ATP entering the binding pocket, the switch-Ⅰ and switch-Ⅱ will cover the ATP molecule and change to the closed state.
, form salt bridges, hydrogen bonds and coordination bonds with the surrounding residues [30,31,32]. N-4 and N-1 form a geometrically matched hydrophobic pocket for the adenosine ring of ATP. The hydrophobic stacking between the pocket and the adenosine ring of ATP molecule contributes to the binding and recognition of nucleotide with the nucleotide-binding pocket of kinesin-1. The entrance of the ATP molecule into the nucleotide-binding pocket can induce a series of conformational changes of the pocket, which can be amplified and finally transmitted to the neck linker portion to initiate the docking movement of neck linker to the motor domain [12, 34,35,36,37,38,39,40,41,42]. The first step of this process is the sensing of the existence of the ATP molecule [43, 44]. The switch-Ⅰ and switch-Ⅱ are the γ-phosphate sensors of kinesin-1. Both the motifs have large conformational changes upon ATP binding into the nucleotide-binding pocket of kinesin family [46]. Comparison of structures of motor domain in different nucleotide-binding states shows that the switch-Ⅰ and switch-Ⅱ are in an open conformation in the nucleotide-free state (apo state). After ATP entering the binding pocket, the switch-Ⅰ and switch-Ⅱ will cover the ATP molecule and change to the closed state.
With the recent progress of experimental method, the high-resolution cryo-EM structures and X-ray crystal structures of kinesin-tubulin complex in different nucleotide-bound states are obtained [45][46][47][48][49][50]. These structures provide much more information about the conformational changes induced by the binding of ATP molecule into the nucleotide-binding pocket. Based on these structures, experimental [47][48][51][52][53] and theoretical [54][55][56] researches confirm that the whole motor domain can be divided into three subdomains. The conformational changes induced by ATP binding can be more exquisitely described as the conformational changes and relative motion of the subdomains. The “alternating cleft” model is proposed on the basis of the above results [48][53]. In this model, there are several clefts on the motor domain in different nucleotide-bound states. In the nucleotide-free state, there is a nucleotide cleft between P-loop (N-1 motif) and switch-Ⅱ (N-3 motif). ATP molecule can enter the binding pocket through this cleft and consequently pull the P-loop ~4 Å toward the microtubule. Interactions between P-loop and switch-Ⅱ in this state can close the nucleotide cleft and induce the rotation of the α2 helix and the central β-sheet to open another cleft, i.e., the “docking cleft”, for the neck linker. Opening of the docking cleft permits the docking movement of neck linker to the motor domain. In the process of nucleotide-cleft closing and docking-cleft opening, the central β-sheet has torsion and twisting [54][48][56]. Distortion of the central β-sheet was analogous to an archery bow by Shang et al. [48]. In the nucleotide-free state, the P-loop and switch-Ⅱ stay apart and the “bow” is in the relaxed state. After ATP binding, interactions between P-loop and switch-Ⅱ result in the distortion of the central β-sheet and initiate the neck-linker docking process. The amphipathic nature of the α4 helix facilitates this subdomain movement [57].
With the recent progress of experimental method, the high-resolution cryo-EM structures and X-ray crystal structures of kinesin-tubulin complex in different nucleotide-bound states are obtained [61,62,66,67,68,69]. These structures provide much more information about the conformational changes induced by the binding of ATP molecule into the nucleotide-binding pocket. Based on these structures, experimental [66,67,70,71,72] and theoretical [33,73,74] researches confirm that the whole motor domain can be divided into three subdomains. The conformational changes induced by ATP binding can be more exquisitely described as the conformational changes and relative motion of the subdomains. The “alternating cleft” model is proposed on the basis of the above results [67,72]. In this model, there are several clefts on the motor domain in different nucleotide-bound states. In the nucleotide-free state, there is a nucleotide cleft between P-loop (N-1 motif) and switch-Ⅱ (N-3 motif). ATP molecule can enter the binding pocket through this cleft and consequently pull the P-loop ~4 Å toward the microtubule. Interactions between P-loop and switch-Ⅱ in this state can close the nucleotide cleft and induce the rotation of the α2 helix and the central β-sheet to open another cleft, i.e., the “docking cleft”, for the neck linker. Opening of the docking cleft permits the docking movement of neck linker to the motor domain. In the process of nucleotide-cleft closing and docking-cleft opening, the central β-sheet has torsion and twisting [33,67,74]. Distortion of the central β-sheet was analogous to an archery bow by Shang et al. [67]. In the nucleotide-free state, the P-loop and switch-Ⅱ stay apart and the “bow” is in the relaxed state. After ATP binding, interactions between P-loop and switch-Ⅱ result in the distortion of the central β-sheet and initiate the neck-linker docking process. The amphipathic nature of the α4 helix facilitates this subdomain movement [75].
In 1999, Rice et al. [9] proved that the conformational change of the neck linker region is a key step in the “walking” process of kinesin-1. Subsequent experimental and theoretical researches [10][12][34][58][59][60][61] showed that the conformational change of the neck linker from undocked state to the docked state can pull the other head to its next binding site on microtubule (the relative position of the two motor domains is shown in Figure 1 and the walking pattern of kinesin-1 is shown in Figure 2). In this way, kinesin-1 “walks” one step on microtubule lattice (the hand-over-hand manner) [62][63][64][65][66][67]. The neck linker of kinesin-1 consists of 14 residues and can be divided into three parts according to their different conformational changes in the docking movement. From the N-terminal end of neck linker, the first part is three residues (Lys323, Thr324 and Ile325, amino acid sequence of 1MKJ [12]), which connect to the α6 helix of the motor domain. After these three residues is the β9 portion of neck linker. In the docked state of the neck linker, the β9 can form a β-sheet with the motor domain. The β9 consists of six residues, Lys326, Asn327, Thr328, Val329, Cys330 and Val331. The C-terminal end of the neck linker is the β10 portion. Five residues, Asn332, Val333, Glu334, Leu335 and Thr336 constitute the β10, which, similar to β9, forms a β-sheet with the motor domain in the docked state. The docking of the neck linker to the motor domain is realized through interactions of these residues with the motor domain [68]. However, the docking mechanisms of these three parts are different. The first step of the neck linker docking is the large conformational change of the three residues on the N-terminal end. In the docked conformation of the neck linker, the three residues (Lys323, Thr324 and Ile325) form a half turn of α-helix structure along the C-terminal end of the α6 helix. This structure formed by the three residues is called the “extra turn” [69][70]. Formation of the extra turn is induced by the rotation of the central β-sheet. This step initiates the subsequent docking of the β9 to the motor domain. The main driving force of β9 docking movement comes from the bending of a special structure formed by β0 and β9, i.e., the “cover-neck bundle” (CNB) [71]. The function of CNB structure was firstly proposed by Hwang et al. [71]. In the MD simulations of Hwang et al. [71], the CNB structure has the spontaneous tendency of bending to the motor domain. This result is also proved by the subsequent experimental research [72]. It is worthy to note that, this CNB mechanism is shared by Eg5 protein, which belongs to the kinesin-5 subfamily [40][73][74]. The CNB-induced neck linker docking may be a universal mechanism applied to the majority of the kinesins. To date, the mechanism of the third step of the neck linker docking, the docking of the β10 to the motor domain, is still unclear. Hwang et al. [71] proposed that the key event of the β10 docking should be the formation of a backbone hydrogen bond between Asn332 and Gly76. They call this hydrogen bond as “Asn latch”. The high strength of this part originates from the cooperation of the hydrophobic residues around the “Asn latch”, which effectively protect the backbone hydrogen bond of the “Asn latch” [75].
In 1999, Rice et al. [9] proved that the conformational change of the neck linker region is a key step in the “walking” process of kinesin-1. Subsequent experimental and theoretical researches [10,12,35,77,78,79, and reviewed in 80] showed that the conformational change of the neck linker from undocked state to the docked state can pull the other head to its next binding site on microtubule (the relative position of the two motor domains is shown in Figure 1 and the walking pattern of kinesin-1 is shown in Figure 2). In this way, kinesin-1 “walks” one step on microtubule lattice (the hand-over-hand manner) [81,82,83,84,85,86]. The neck linker of kinesin-1 consists of 14 residues and can be divided into three parts according to their different conformational changes in the docking movement. From the N-terminal end of neck linker, the first part is three residues (Lys323, Thr324 and Ile325, amino acid sequence of 1MKJ [12]), which connect to the α6 helix of the motor domain. After these three residues is the β9 portion of neck linker. In the docked state of the neck linker, the β9 can form a β-sheet with the motor domain. The β9 consists of six residues, Lys326, Asn327, Thr328, Val329, Cys330 and Val331. The C-terminal end of the neck linker is the β10 portion. Five residues, Asn332, Val333, Glu334, Leu335 and Thr336 constitute the β10, which, similar to β9, forms a β-sheet with the motor domain in the docked state. The docking of the neck linker to the motor domain is realized through interactions of these residues with the motor domain [87]. However, the docking mechanisms of these three parts are different. The first step of the neck linker docking is the large conformational change of the three residues on the N-terminal end. In the docked conformation of the neck linker, the three residues (Lys323, Thr324 and Ile325) form a half turn of α-helix structure along the C-terminal end of the α6 helix. This structure formed by the three residues is called the “extra turn” [88, 89]. Formation of the extra turn is induced by the rotation of the central β-sheet. This step initiates the subsequent docking of the β9 to the motor domain. The main driving force of β9 docking movement comes from the bending of a special structure formed by β0 and β9, i.e., the “cover-neck bundle” (CNB) [90]. The function of CNB structure was firstly proposed by Hwang et al. [90]. In the MD simulations of Hwang et al. [90], the CNB structure has the spontaneous tendency of bending to the motor domain. This result is also proved by the subsequent experimental research [91]. It is worthy to note that, this CNB mechanism is shared by Eg5 protein, which belongs to the kinesin-5 subfamily [41,92,93]. The CNB-induced neck linker docking may be a universal mechanism applied to the majority of the kinesins. To date, the mechanism of the third step of the neck linker docking, the docking of the β10 to the motor domain, is still unclear. Hwang et al. [90] proposed that the key event of the β10 docking should be the formation of a backbone hydrogen bond between Asn332 and Gly76. They call this hydrogen bond as “Asn latch”. The high strength of this part originates from the cooperation of the hydrophobic residues around the “Asn latch”, which effectively protect the backbone hydrogen bond of the “Asn latch” [94].
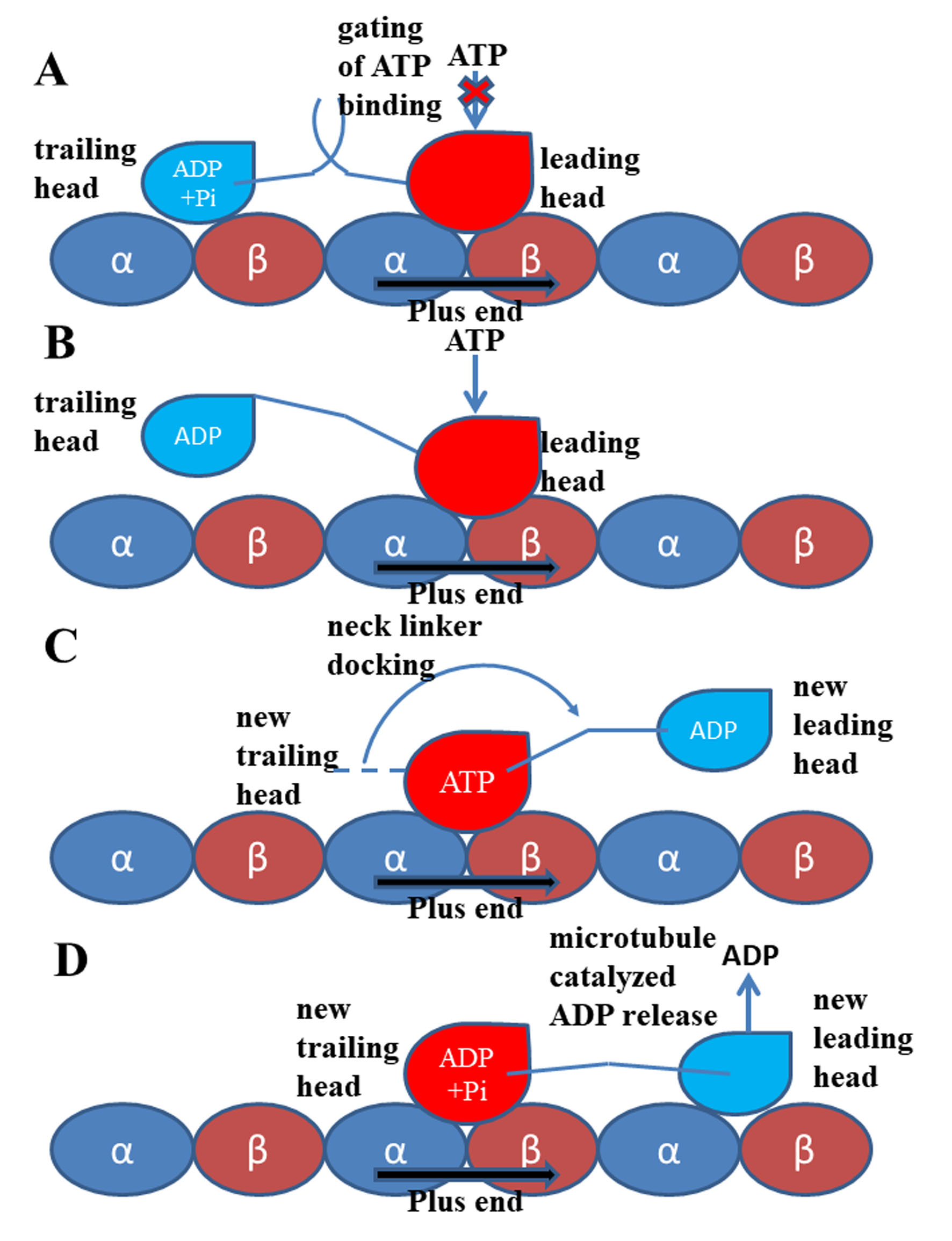
Figure 2.
Walking and mechanochemical coupling mechanisms of kinesin-1. (A) Both motor domains bind to microtubule strongly (same with Figure 1). In this state, the internal strain between the two motor domains and the backward orientated neck linker of the leading head inhibit the binding of ATP molecule to this motor domain. (B) Detachment of the trailing head from the microtubule releases the internal strain and the restriction to the orientation of the neck linker of the leading head. The ATP molecule can bind to the leading head. (C) The neck linker docking induced by the ATP binding pulls the trailing head to the next binding site on microtubule. (D) The ADP-bound state new leading head binds to microtubule. The ADP molecule releases from this head quickly due to the catalysis of the microtubule.
3. Perspective
3.Perspective
After ~35 years of investigation, the walking mechanism and the mechanochemical coupling of kinesin-1 is much clearer now. The energy for mechanical walking of kinesin-1 comes from ATP. Because the ATP molecule is charged, the binding of ATP molecule into the nucleotide-binding site on the motor domain of kinesin-1 can induce a series of conformational changes of the motor domain. These conformational changes are finally transmitted to the neck linker and, thus, trigger the docking movement of the neck linker. However, there are still some problems unresolved. For example, how the β10 docking is accomplished in the neck linker docking process? In vivo, the kinesin-1 proteins often interact with some microtubule associated proteins or other proteins to work effectively [76]. To transport large organelles along the microtubules, the kinesin-1 proteins often cooperate to achieve a high moving speed. Besides the regulation of the ADP release of kinesin-1 by the microtubule, the posttranslational modifications of the microtubules can also affect the activity of the kinesin-1 [77][78][79]. The detailed mechanisms of all these aspects at the residue level are still unknown and we still have a long way to go to achieve a complete understanding of kinesin mechanism.
After ~35 years of investigation, the walking mechanism and the mechanochemical coupling of kinesin-1 is much clearer now. The energy for mechanical walking of kinesin-1 comes from ATP. Because the ATP molecule is charged, the binding of ATP molecule into the nucleotide-binding site on the motor domain of kinesin-1 can induce a series of conformational changes of the motor domain. These conformational changes are finally transmitted to the neck linker and, thus, trigger the docking movement of the neck linker. However, there are still some problems unresolved. For example, how the β10 docking is accomplished in the neck linker docking process? In vivo, the kinesin-1 proteins often interact with some microtubule associated proteins or other proteins to work effectively [130]. To transport large organelles along the microtubules, the kinesin-1 proteins often cooperate to achieve a high moving speed. Besides the regulation of the ADP release of kinesin-1 by the microtubule, the posttranslational modifications of the microtubules can also affect the activity of the kinesin-1 [131,132,133]. The detailed mechanisms of all these aspects at the residue level are still unknown and we still have a long way to go to achieve a complete understanding of kinesin mechanism.