In biosciences and biotechnologies, it is recently critical to promote research regarding the regulation of the dynamic functions of proteins of interest. Light-induced control of protein activity is a strong tool for a wide variety of applications because light can be spatiotemporally irradiated in high resolutions. Therefore, synthetic, semi-synthetic, and genetic engineering techniques for photoactivation of proteins have been actively developed. In this review, the conventional approaches will be outlined. As a solution for overcoming barriers in conventional ones, ourresearchers' recent approaches in which proteins were chemically modified with biotinylated caging reagents are introduced to photo-activate a variety of proteins without genetic engineering and elaborate optimization.
- protein caging
- optogenetics
- genetic code extension technology
- expressed protein ligation
- photolytic protein aggregates
1. Introduction
2. Caged Proteins
Methods for converting proteins to photo-responsive ones by reversible chemical modification have also been used as a complementary approach in which intact proteins with no modification are produced after photoactivation [10][11][12][21][22][23]. In the field of chemical biology, inactivation of biomolecules by modification with photodegradable protective groups is called “caging”, and such protected compounds are called caged compounds [10][21]. Caging have been employed for activating biomolecules of interest at desired timing and location by exposure to light (Figure 1A). For example, a caged glutamine was reported to be introduced into a neuron, and then one of the dendrites was site-specifically exposed to light. As a result, the shape change of the light-exposed dendrites clearly indicated the molecular system in which glutamine affects the shape memory of neural spines [24]. Thus, caging has been utilized as a powerful research tool for giving new insight into molecular mechanisms in cell biology. Such a caging approach has also been applied to proteins, and caged proteins have been reported for more than a quarter of a century [22][23]. The simplest strategy for preparing caged proteins is to randomly introduce a photodegradable protecting group to the amino group on the protein surface through the reaction with an active carbonyl group (Figure 1B) [25][26][27][28]. In this method, in principle, almost all proteins can be caged without any pretreatment simply by mixing a reactive photodegradable protecting reagent (a caging reagent). After the protective group is degraded by light, the caged lysine residue returns to the original lysine one without leaving any trace, so there is no need to consider the effect of caging on the protein function after photoactivation. This is a great advantage when studying the function of a protein or using it as a drug. Actually, the polymerization activity of G-actin was photo-regulated by this caging strategy [25]. The selective binding function of antibodies were demonstrated to be remotely controlled by light exposure [27]. In a similar way, the carboxylic acid moiety on protein surfaces was reported to be randomly blocked with the diazo derivatives of photolytic protection groups [29]. This method realized photoactivation of hemoglobin. However, unlike small caged compounds, such random introduction of a small photodegradable protecting group onto the surface of a large protein often fails to fully inactivate the function of the protein (Figure 1C). In the case of enzymes, for example, this simple strategy based on random amine modification can function only on the enzymes that happen to have lysine residues at sites involved in catalytic activity or substrate binding (Figure 1B). Therefore, caging of proteins often requires the strategy of introducing the photodegradable protecting group specifically at the sites involved in their activity.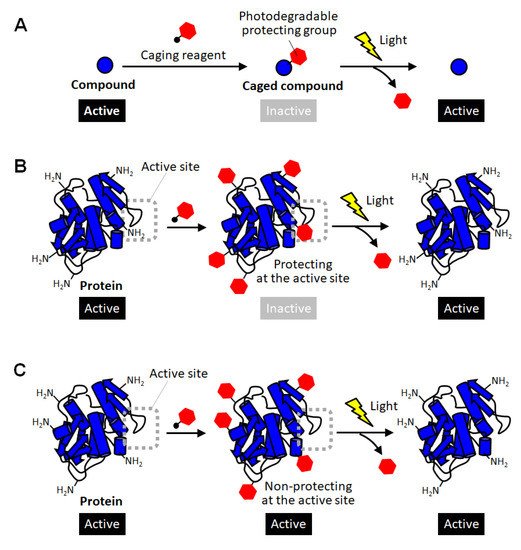
3. Site-Specific Protein Caging
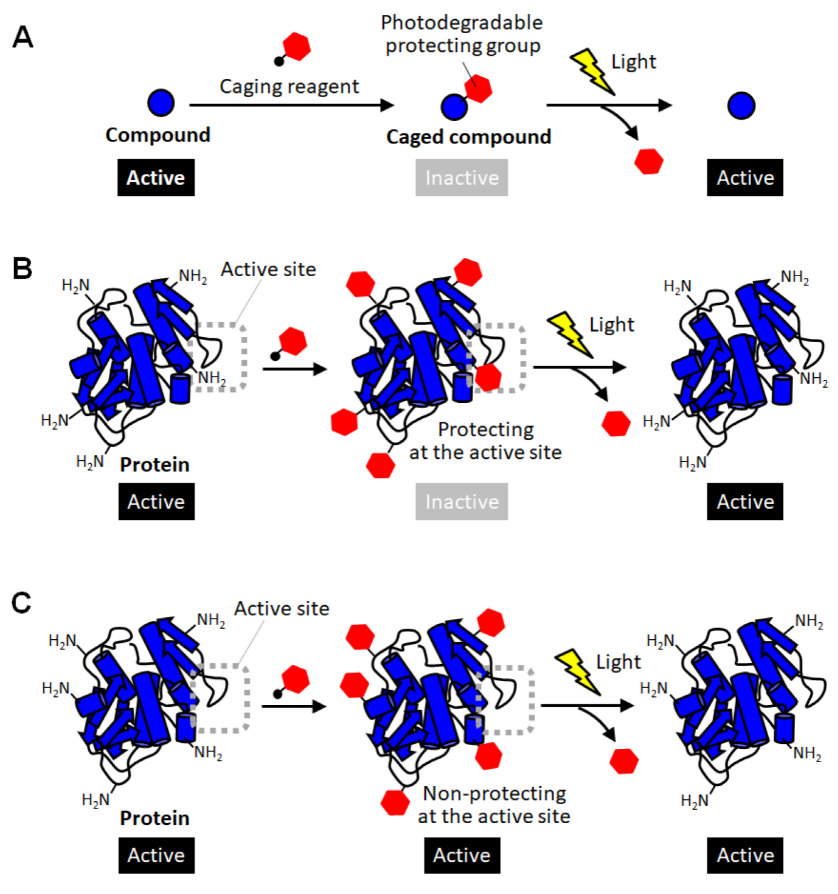
As a method for site-specific introduction of the photodegradable protecting group, the use of the cysteine residue of the proteins has been utilized [11][30][31][32][33][34]. The selective reactivity of the thiol group realized the site-specific caging of the cysteine residue of which there are often one or a few on protein surfaces. Similar to the random amine coupling, the function of intact proteins was reported to be photo-regulated by caging with thiol-reactive protecting reagents when the cysteine residues were at the sites involved in protein functions [30][31]. By this approach, the enzymatic activities of myosin [30] and β-galactosidase [31] were demonstrated to be activated by light exposure. As another method using the thiol-based reaction, the caging of thiophosphorylated proteins was also reported [32]. In this method, the threonine residue of a protein was thiophosphorylated using 3-phosphoinositide-dependent kinase, and then modified with a thiol-reactive caging reagent. After photolysis, the caged protein converted to the thiophosphorylated protein which exerted almost the same activity as the phosphorylated one. This kinase-coupled approach may be versatile for caging a variety of phosphorylated proteins. To apply such thiol-based site-specific caging approaches to any other proteins, the use of cysteine-substituted mutants has been widely utilized (Figure 2A) [11][33][34]. In this approach, the function-related site is replaced with cysteine, and a caging reagent that selectively reacts with the thiol group is applied. Based on this approach, the actin polymerization activity of cofilin, which was unknown so far, was exerted by light exposure in living cells, giving new insights into the role of cofilin [11]. Furthermore, light-induced depolymerization of actin was also achieved by caged cofilin on the glass substrate [33], and the pore-formation on cellular membranes was photo-induced by caged α-hemolysin [34]. Although this approach is simple to introduce a protecting group at the desired active site, the proteins produced after photolysis are only the cysteine-substituted mutant protein on which the mutation is introduced at a critical site involved in function (Figure 2A). Therefore, there is a high risk that the light-induced activity of the mutant protein will be inferior to that of the native one. Thus, the caging strategy based on post-translational chemical modification often requires the site-specific replacement with reactive amino acids for selective caging, and therefore, almost exclusively applied to mutant proteins.
References
- Gautier, A.; Gauron, C.; Volovitch, M.; Bensimon, D.; Jullien, L.; Vriz, S. How to control proteins with light in living systems. Nat. Chem. Biol. 2014, 10, 533–541.
- Wu, Y.I.; Frey, D.; Lungu, O.I.; Jaehrig, A.; Schlichting, I.; Kuhlman, B.; Hahn, K.M. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 2009, 461, 104–108.
- Inagaki, H.K.; Jung, Y.; Hoopfer, E.D.; Wong, A.M.; Mishra, N.; Lin, J.Y.; Tsien, R.Y.; Anderson, D.J. Optogenetic control of Drosophila using a red-shifted channel rhodopsin reveals experience-dependent influences on courtship. Nat. Methods 2014, 11, 325–332.
- Morri, M.; Sanchez-Romero, I.; Tichy, A.M.; Kainrath, S.; Gerrard, E.J.; Hirschfeld, P.P.; Schwarz, J.; Janovjak, H. Optical functionalization of human Class A orphanG-protein-coupled receptors. Nat. Commun. 2018, 9, 1950.
- Wang, J.; Liu, Y.; Liu, Y.; Zheng, S.; Wang, X.; Zhao, J.; Yang, F.; Zhang, G.; Wang, C.; Chen, P.R. Time-resolved protein activation by proximal decaging in living systems. Nature 2019, 569, 509–513.
- Tomatsu, I.; Peng, K.; Kros, A. Photoresponsive hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2011, 63, 1257–1266.
- Høgset, A.; Prasmickaite, L.; Selbo, P.K.; Hellum, M.; Engesæter, B.Ø.; Bonsted, A.; Berg, K. Photochemical internalisation in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 95–115.
- Gu, Z.; Biswas, A.; Zhao, M.; Tang, Y. Tailoring nanocarriers for intracellular protein delivery. Chem. Soc. Rev. 2011, 40, 3638–3655.
- Sasaki, Y.; Akiyoshi, K. Nanogel engineering for new nanobiomaterials: From chaperoning engineering to biomedical applications. Chem. Rec. 2010, 10, 366–376.
- Brieke, C.; Rohrbach, F.; Gottschalk, A.; Mayer, G.; Heckel, A. Light-controlled tools. Angew. Chem. Int. Ed. 2012, 51, 8446–8476.
- Ghosh, M.; Song, X.; Mouneimne, G.; Sidani, M.; Lawrence, D.S.; Condeelis, J.S. Cofilin promotes actin polymerization and defines the direction of cell motility. Science 2004, 304, 743–746.
- Deiters, A.; Groff, D.; Ryu, Y.; Xie, J.; Schultz, P.G. A genetically encoded photocaged tyrosine. Angew. Chem. Int. Ed. 2006, 45, 2728–2731.
- Selbo, P.K.; Sandvig, K.; Kirveliene, V.; Berg, K. Release of gelonin from endosomes and lysosomes to cytosol by photochemical internalization. Biochim. Biophys. Acta 2000, 1475, 307–313.
- Peng, K.; Tomatsu, I.; Kros, A. Light controlled protein release from a supramolecular hydrogel. Chem. Commun. 2010, 46, 4094–4096.
- Levskaya, A.; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 2009, 461, 997–1001.
- Kim, N.; Kim, J.M.; Lee, M.; Kim, C.Y.; Chang, K.Y.; Heo, W.D. Spatiotemporal control of fibroblast growth factor receptor signals by blue light. Chem. Biol. 2014, 21, 903–912.
- Nihongaki, Y.; Kawano, F.; Nakajima, T.; Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015, 33, 755–760.
- Gil, A.A.; Carrasco-López, C.; Zhu, L.; Zhao, E.M.; Ravindran, P.T.; Wilson, M.Z.; Goglia, A.G.; Avalos, J.L.; Toettcher, J.E. Optogenetic control of protein binding using light-switchable nanobodies. Nat. Commun. 2020, 11, 4044.
- Zhang, W.; Lohman, A.W.; Zhuravlova, Y.; Lu, X.; Wiens, M.D.; Hoi, H.; Yaganoglu, S.; Mohr, M.A.; Kitova, E.N.; Klassen, J.S.; et al. Optogenetic control with a photocleavable protein, Phocl. Nat. Methods 2017, 14, 391–394.
- Lu, X.; Wen, Y.; Zhang, S.; Zhang, W.; Chen, Y.; Shen, Y.; Lemieux, M.J.; Campbell, R.E. Photocleavable proteins that undergo fast and efficient dissociation. Chem. Sci. 2021, 12, 9658–9672.
- Ellis-Davies, G.C.R. Caged compounds: Photorelease technology for control of cellular chemistry and physiology. Nat. Methods 2007, 4, 619–6289.
- Curley, K.; Lawrence, D.S. Light-activated proteins. Curr. Opin. Chem. Biol. 1999, 3, 84–88.
- Lawrence, D.S. The preparation and in vivo applications of caged peptides and proteins. Curr. Opin. Chem. Biol. 2005, 9, 570–575.
- Matsuzaki, M.; Ellis-Davies, G.C.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001, 4, 1086–1092.
- Marriott, G. Caged protein conjugates and light-directed generation of protein activity: Preparation, photoactivation, and spectroscopic characterization of caged G-actin conjugates. Biochemistry 1994, 33, 9092–9097.
- Thompson, S.; Spoors, J.A.; Fawcett, M.-C.; Self, C.H. Photocleavable nitrobenzyl–protein conjugates. Biochem. Biophys. Res. Commun. 1994, 201, 1213–1219.
- Self, C.H.; Thompson, S. Light activatable antibodies: Models for remotely activatable proteins. Nat. Med. 1996, 2, 817–820.
- Ottl, J.; Gabriel, D.; Marriott, G. Preparation and photoactivation of caged fluorophores and caged proteins using a new class of heterobifunctional, photocleavable cross-linking reagents. Bioconj. Chem. 1998, 9, 143–151.
- Bédouet, L.; Adenier, H.; Pulvin, S.; Bedel-Cloutour, C.; Thomas, D. Recovery of the oxidative activity of caged bovine haemoglobin after UV photolysis. Biochem. Biophys. Res. Commun. 2004, 320, 939–944.
- Marriot, G.; Heidecker, M. Light-directed generation of the actin activated ATPase activity of caged heavy meromysin. Biochemistry 1996, 35, 3170–3174.
- Golan, R.; Zehavi, U.; Naim, M.; Patchornik, A.; Smirnoff, P.; Inhibition of, E. coli β-galactosidase by 2-nitro-1-(4,5-dimethoxy-2-nitrophenyl)ethyl, a photo-reversible thiol label. Biochim Biophys Acta 1996, 1293, 238–242.
- Zou, K.; Cheley, S.; Givens, R.S.; Bayley, H. Catalytic subunit of protein kinase A caged at the activating phosphothreonine. J. Am. Chem. Soc. 2002, 124, 8220–8229.
- Ghosh, M.; Ichetovkin, I.; Song, X.; Condeelis, J.S.; Lawrence, D.S. A new strategy for caging proteins regulated by kinases. J. Am. Chem. Soc. 2002, 124, 2440–2441.
- Chang, C.Y.; Niblack, B.; Walker, B.; Bayley, H. A photogenerated pore-forming protein. Chem. Biol. 1995, 2, 391–400.
- Mendel, D.; Eilman, J.A.; Schultz, P.G. Construction of a light-activated protein by unnatural amino acid mutagenesis. J. Am. Chem. Soc. 1991, 113, 2758–2760.
- Cook, S.N.; Jack, W.E.; Xiong, X.; Danley, L.E.; Ellman, J.A.; Schultz, P.G.; Noren, C.J. Photochemically lnitiated Protein Splicing. Angew. Chem. Int. Ed. 1995, 34, 1629–1630.
- England, P.M.; Lester, H.A.; Davidson, N.; Dougherty, D.A. Site-specific, photochemical proteolysis applied to ion channels in vivo. Proc. Natl. Acad. Soc. USA 1997, 94, 11025–11030.
- Miller, J.C.; Silverman, S.K.; England, P.M.; Dougherty, D.A.; Lester, H.A. Flash decaging of tyrosine sidechains in an ion channel. Neuron 1998, 20, 619–624.
- Short, G.F.; Lodder, M.; Laikhter, A.L.; Arslan, T.; Hecht, S.M. Caged HIV-1 protease: Dimerization is independent of the ionization state of the active site aspartates. J. Am. Chem. Soc. 1999, 121, 478–479.
- Tong, Y.; Brandt, G.S.; Li, M.; Shapovalov, G.; Slimko, E.; Karschin, A.; Dougherty, D.A.; Lester, H.A. Tyrosine decaging leads to substantial membrane trafficking during modulation of an inward rectifier potassium channel. J. Gen. Physiol. 2001, 117, 103–118.
- Petersson, E.J.; Brandt, G.S.; Zacharias, N.M.; Dougherty, D.A.; Lester, H.A. Caging proteins through unnatural amino acid mutagenesis. Methods Enzymol. 2003, 360, 258–273.
- Wu, N.; Deiters, A.; Cropp, T.A.; King, D.; Schultz, P.G. A genetically encoded photocaged amino acid. J. Am. Chem. Soc. 2004, 126, 14306–14307.
- Endo, M.; Nakayama, K.; Kaida, Y.; Majima, T. Design and synthesis of photochemically controllable caspase-3. Angew. Chem. Int. Ed. 2004, 116, 5761–5763.
- Endo, M.; Nakayama, K.; Majima, T. Design and synthesis of photochemically controllable restriction endonuclease BamHI by manipulating the salt-bridge network in the dimer interface. J. Org. Chem. 2004, 69, 4292–4298.
- Cornish, V.W.; Mendel, D.; Schultz, P.G. Probing protein structure and function with an expanded genetic code. Angew Chem Int Ed 1995, 34, 621–633.
- Rothman, D.M.; Petersson, E.J.; Vázquez, M.E.; Brandt, G.S.; Dougherty, D.A.; Imperiali, B. Caged phosphoproteins. J. Am. Chem. Soc. 2005, 127, 846–847.
- Ren, W.; Ji, A.; Ai, H. Caging proteins through unnatural amino acid mutagenesis. J. Am. Chem. Soc. 2015, 137, 2155–2158.
- Edwards, W.F.; Young, D.D.; Deiters, A. Light-activated cre recombinase as a tool for the spatial and temporal control of gene function in mammalian cells. ACS. Chem. Biol. 2009, 4, 441–445.
- Gautier, A.; Nguyen, D.P.; Lusic, H.; An, W.; Deiters, A.; Chin, J.W. Genetically encoded photocontrol of protein localization in mammalian cells. J. Am. Chem. Soc. 2010, 132, 4086–4088.
- Arbely, E.; Torres-Kolbus, J.; Deiters, A.; Chin, J.W. Photocontrol of tyrosine phosphorylation in mammalian cells via genetic encoding of photocaged tyrosine. J. Am. Chem. Soc. 2012, 134, 11912–11915.
- Lemke, E.A.; Summerer, D.; Geierstanger, B.H.; Brittain, S.M.; Schultz, P.G. Control of protein phosphorylation with a genetically encoded photocaged amino acid. Nat. Chem. Biol. 2007, 3, 769–772.
- Walker, O.S.; Elsässer, S.J.; Mahesh, M.; Bachman, M.; Balasubramanian, S.; Chin, J.W. Photoactivation of mutant isocitrate dehydrogenase 2 reveals rapid cancer-associated metabolic and epigenetic changes. J. Am. Chem. Soc. 2016, 138, 718–721.
- Hemphill, J.; Borchardt, E.K.; Brown, K.; Asokan, A.; Deiters, A. Optical Control of CRISPR/Cas9 Gene Editing. J. Am. Chem. Soc. 2015, 137, 5642–5645.
- Hiraoka, T.; Hamachi, I. Caged RNase: Photoactivation of the enzyme from perfect off-state by site-specific incorporation of 2-nitrobenzyl moiety. Bioorg. Med. Chem. Lett. 2003, 13, 13–15.
- Pellois, J.P.; Hahn, M.E.; Muir, T.W. Simultaneous triggering of protein activity and fluorescence. J. Am. Chem. Soc. 2004, 126, 7170–7171.
- Hahn, M.E.; Muir, T.W. Photocontrol of Smad2, a multiphosphorylated cell-signaling protein, through caging of activating phosphoserines. Angew. Chem. Int. Ed. 2004, 43, 5800–5803.
- Cotton, G.J.; Muir, T.W. Peptide ligation and its application to protein engineering. Chem. Biol. 1999, 6, R247–R256.
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1999, 266, 776–778.
- Tang, S.; Wan, Z.; Gao, Y.; Zheng, J.S.; Wang, J.; Si, Y.Y.; Chen, X.; Qi, H.; Liu, L.; Liu, W. Total chemical synthesis of photoactivatable proteins for light-controlled manipulation of antigen–antibody interactions. Chem. Sci. 2016, 7, 1891–1895.
- Curley, K.; Lawrence, D.S. Photoactivation of a signal transduction pathway in living cells. J. Am. Chem. Soc. 1998, 120, 8573–8574.
- Loudwig, S.; Nicolet, Y.; Masson, P.; Fontecilla-Camps, J.C.; Bon, S.; Nachon, F.; Goeldner, M. Photoreversible inhibition of cholinesterases: Catalytic serine-labeled caged butyrylcholinesterase. ChemBioChem 2003, 4, 762–767.
- Tsukiji, S.; Miyagawa, M.; Takaoka, Y.; Tamura, T.; Hamachi, I. Ligand-directed tosyl chemistry for protein labeling in vivo. Nat. Commun. 2008, 5, 341–343.
- Fujishima, S.; Yasui, R.; Miki, T.; Ojida, A.; Hamachi, I. Ligand-directed acyl imidazole chemistry for labeling of membrane-bound proteins on live cells. J. Am. Chem. Soc. 2012, 134, 3961–3964.
- Tamura, T.; Ueda, T.; Goto, T.; Tsukidate, T.; Shapira, Y.; Nishikawa, Y.; Fujisawa, A.; Hamachi, I. Rapid labelling and covalent inhibition of intracellular native proteins using ligand-directed N-acyl-N-alkyl sulfonamide. Nat. Commun. 2018, 14, 1870.
- Matsuo, K.; Kioi, Y.; Yasui, R.; Takaoka, Y.; Miki, T.; Fujishima, S.; Hamachi, I. One-step construction of caged carbonic anhydrase I using a ligand-directed acyl imidazole-based protein labeling method. Chem. Sci. 2013, 4, 2573–2580.