Chloroquine (CQ) and hydroxychloroquine (HCQ) have been proposed as treatments for COVID-19. These drugs have been studied for many decades, primarily in the context of their use as antimalarials, where they induce oxidative stress-killing of the malarial parasite. Less appreciated, however, is evidence showing that CQ/HCQ causes systemic oxidative stress. In vitro and observational data suggest that CQ/HCQ can be repurposed as potential antiviral medications.
Potential adverse affects of CQ/HCQ on COVID-19 patients. CQ/HCQ induced oxidative stress could contribute to the pathology of acute respiratory distress syndrome (ARDS) and hypoxia. Hypoxia, in turn, contribute to increased production of ROS from mitochondria (see text). CQ/HCQ induced damage to type II alveolar cells (T2ACs) and surfactant function could also contribute to ARDS and hypoxia/hypoxemia. As indicated in Figure 4, CQ can form a complex with free heme that promotes biomembrane lipid peroxidation and this process could contribute to the systemic coagulopathy found in severe COVID-19 (see text). CQ/HCQ could also alter the immune responsive by promoting a shift in macrophage phenotype M2 to the M1 (see text). The M1 macrophage phenotype is proinflammatory and promotes tissue damage. The potential adverse affects of CQ/HCQ summarized in this Figure need to be further confirmed by additional in vivo and in vitro studies,
- chloroquine
- hydroxychloroquine
- proton fluxes
- COVID-19
- oxidative stress
- cytokine storm
- SARS-CoV-2
- macrophage
- immune response
- reactive oxygen species
1. Introduction
Definition
Chloroquine (CQ) and hydroxychloroquine (HCQ) have been proposed as treatments for COVID-19. These drugs have been studied for many decades, primarily in the context of their use as antimalarials, where they induce oxidative stress-killing of the malarial parasite. Less appreciated, however, is evidence showing that CQ/HCQ causes systemic oxidative stress. In vitro and observational data suggest that CQ/HCQ can be repurposed as potential antiviral medications.
1. Introduction
At the writing of this work (mid-2020), there was no Food and Drug Administration (FDA) approved drugs for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA virus infection, which causes the coronavirus disease 2019 (COVID-19). Both chloroquine (CQ) and hydroxychloroquine (HCQ) are 4-aminoquinoline drugs with similar structures, as shown in
Figure 1. CQ and HCQ have been proposed as potential COVID-19 medical countermeasures
. CQ and HCQ have been proposed as potential COVID-19 medical countermeasures [
[1][2][3][4][5][6]. The FDA currently approves CQ/HCQ for use as antimalarials, the treatment of rheumatoid arthritis, and systemic lupus erythematosus (SLE) where they act by immunomodulatory mechanisms [7]. HCQ reduces disease activity in SLE patients but has no significant effect on pro-inflammatory cytokines [8]. A recent literature review highlights many immunomodulatory mechanisms influenced by CQ/HCQ and emphasizes our lack of current knowledge concerning their potential effects on immune responses in COVID-19 patients [9]. CQ/HCQ use may lead to unknown alterations in immune responses in COVID-19 patients, including diminished innate immune responses as well as potential modifications of the B and T cell responses to the COVID-19 virus [9].
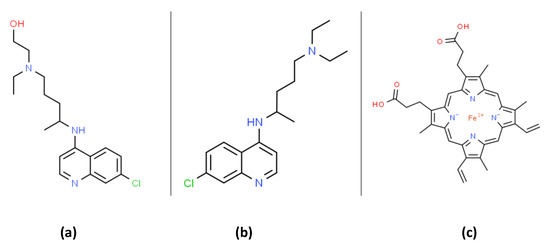
Figure 1. The organic structures of (a) hydroxychloroquine (HCQ); (b) chloroquine and; (c) heme. Chloroquine can form a membrane bound complex with heme that can promote lipid peroxidation.
Both in vitro and in vivo animal experiments have demonstrated the anti-coronavirus activity of CQ /HCQ [10][11][12]. Moreover, CQ/HCQ are broadly available, cost-effective, and therefore attractive potential therapies for viral pandemics without an effective vaccine. A review by Cortegiani et al. on the efficacy and safety of CQ for treating COVID-19 concluded that available pre-clinical evidence was sufficiently robust to justify initiating high-quality clinical trials [4]. Based on available data, the FDA initially published an emergency use authorization of CQ/HCQ (on 28 March 2020) for COVID-19 treatment. The FDA subsequently revoked this emergency use authorization (on 15 June 2020) due to new clinical data suggesting CQ/HCQ were ineffective and raising concerns regarding "serious cardiac adverse events and other potential serious side effects" [13]. Nevertheless, the FDA states "additional clinical trials continue to evaluate the potential benefit of these drugs in treating or preventing COVID-19. "The molecular basis for the potential antiviral activity of CQ/HCQ are not fully understood, and multiple mechanisms have been proposed [9]. This review will explore the hypothesis that alterations in proton fluxes and redox physiology induced by CQ/HCQ are relevant to their potential antiviral activity and side-effects. The production of reactive oxygen species (ROS) and cellular/subcellular proton fluxes are interdependent factors modulating many antimicrobial immune responses [14]. We will also highlight concerns that CQ/HCQ-induced oxidative stress could be problematic in the treatment of critically ill COVID-19 patients. The "cytokine storm" occurring in some severe forms of viral infection (e.g., influenza A viruses) is associated with increased oxidative stress as well as increased morbidity and mortality [15]. Accumulating evidence suggests that this cytokine storm is a significant cause of ARDS and multiorgan failure in COVID-19 patients [16]. Considerable evidence suggests that parasite-specific oxidative stress induced by CQ/HCQ treatment accounts for the antimalarial activity of these drugs [17]. Less appreciated, however, is evidence (see below) showing that CQ/HCQ, by themselves, are pro-oxidants that can increase oxidative stress parameters [18][19][20][21]. Viral infections are also generally accompanied by oxidative stress with potential pathophysiological consequences [22]. As mentioned above, compelling evidence supports the view that patients with COVID-19 are at increased risk of developing ARDS and subsequent death from respiratory failure [23]. Decades of research have demonstrated the central role of oxidative stress in ARDS pathophysiology [24][25][26][27]. Understanding the potential role of oxidative stress in COVID-19 is critical, since potential anti-COVID-19 drug candidates that increase oxidative stress have the potential to exacerbate ARDS pathophysiology.
2. CQ/HCQ Pharmacology and Alterations in Subcellular Organelles Proton Fluxes
Before reviewing the role of CQ/HCQ in redox physiology and SARS-CoV-2 infection, we will briefly summarize the basics of CQ/HCQ pharmacology (see Figure 2) with an emphasis on CQ/HCQ-induced alterations in subcellular organelle proton fluxes. CQ/HCQ-induced changes in proton fluxes will have a direct effect on the type of ROS present, and thereby influence their potential effects on redox-sensitive signal transduction pathways and potential molecular and cellular damage. Among the key redox-sensitive pathways are those controlled by transcription factor nuclear factor erythroid 2 p45-related factor 2 (NRF2), hypoxia-inducible transcription factors (HIFs), nuclear factor-κB (NF-κB) transcription factors, and activator protein-1 (AP-1) transcription factors (see below as well). Excellent, comprehensive reviews of CQ/HCQ pharmacokinetics and their side-effects are available [9][28][29].
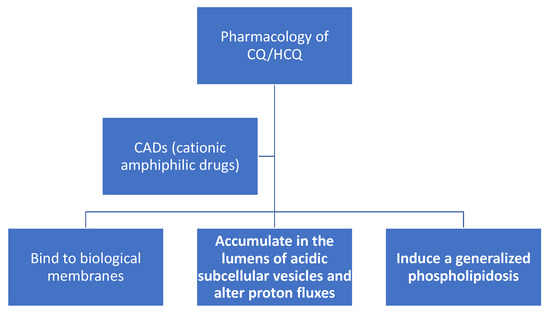
Figure 2. Chloroquine (CQ) and hydroxychloroquine (HCQ) are cationic amphiphilic drugs (CADs) that share a set of common pharmacological properties: (a) they can partition into biomembranes; (b) accumulate in the lumens of acidic subcellular organelles, and; (c) induce a generalized phospholipidosis, which is the lysosomal accumulation of phospholipids.
3. CQ/HCQ Bind to Biological Membranes and Alters their Structure/Function
After many decades of research, some of the least controversial characteristics of CQ/HCQ are the ability of these weakly basic and lipophilic compounds: (1) to bind biological membranes; (2) to accumulate in the lumens of acidic subcellular vesicles and alter proton fluxes; (3) to induce generalized cellular phospholipidosis. These effects are relevant to the production of ROS, proton fluxes and the immunomodulatory and potential antiviral activities of CQ/HCQ.
CQ and HCQ both belong to a class of compounds (see Figure 1 and Figure 2) termed cationic amphiphilic drugs or CADs [30]. In plasma (pH 7.4) and cellular cytoplasm (pH 7.2), the divalent forms of CQ/HCQ are the dominant species. At pH 7.4 CQ, CQH+ (monovalent), and CQH22+ (divalent) are present at 0.026, 16.74, and 83.23%, respectively (reaction 1). Unambiguous in vitro experiments show that CQH22+ binds to the phospholipid bilayers of multilamellar liposomes with a robust partition coefficient [31]. Phosphatidylserine (PS) liposomes, with a negative charge, binds CQH22+ with a particularly high partition coefficient [31]. PS plays a central role in apoptosis. Proton NMR studies have confirmed the binding of CQH22+ to phosphatidylcholine liposomes [32]. As expected, resonance signals from protons on the hydrophobic ring carbons of CADs are affected by association with liposomes [32].

The binding of CADs to lipid bilayers is stabilized by nonspecific hydrophobic and ionic interactions [31]. Lipid bilayers are present in all cells and many subcellular organelles. It follows that CADs can accumulate, to some extent, in the cells and membranous organelles of most tissues. CAD accumulation can potentially affect many functions of all biological membranes by altering their structure and functions, e.g., membrane-bound lysosomal phosphatases and hydrolases [33].
4. CQ/HCQ Accumulate in the Lumens of Acidic Subcellular Vesicles and Alter Proton Fluxes
It is also well established that CADs accumulate in the lumens of acidic subcellular vesicles. The neutral forms of CADs can freely diffuse through the hydrophobic domain of lipid bilayers. For acidified vesicles, such as endosomes, lysosomes, M2 phagosomes, and pulmonary lysosomal lamellar bodies, the neutral forms of CADs will become charged inside the lumen and will no longer be able to diffuse out [28]. Acidified vesicles utilize a vacuolar proton-pumping ATPase (V-ATPase) to maintain a low pH by pumping protons across the vesicular membrane into the lumen [34]. For lysosomes attempting to maintain a low pH (pH 4.8) in the presence of CQ/HCQ, there will be a gradual luminal accumulation of CQ/HCQ, eventually leveling off at levels as high as 20 mM [28][35]. This process is termed "lysosomal trapping," and trapped CADs are denoted as "lysosomotropic" [35][36][37]. CQ shows a wide variation in lysosomal trapping between organs with lungs > kidney = brain = liver > diaphragm = heart = skeletal muscles > adipose tissue [38]. The high CQ accumulation in the lungs is relevant to respiratory distress disorders [39]. Lamellar bodies are a type of acidified lysosome found in type II alveolar cells (T2ACs), and keratinocytes and these organelles are known to accumulate weak bases such as CQ/HCQ [40][41]. The lamellar bodies found in T2ACs secrete surfactant, which is essential for pulmonary alveoli gas exchange [42]. T2ACs are particularly relevant to COVID-19, since these cells express angiotensin-converting enzyme-2 (ACE-2), which SARS-CoV-2 utilizes as a receptor to enter the lungs. T2AC cells are preferentially infected by SARS-CoV-2, potentially contributing to a reduced secretion or function of surfactant with a resulting loss of pulmonary compliance [43]. Reduced pulmonary compliance is a typical characteristic of ARDS, but exogenous surfactant treatment has not proven to be therapeutically effective [44][45]. Nevertheless, there is a pharmaceutical interest in testing surfactant therapy in COVID-19 patients. CQ interferes with the processing of surfactant protein C, which is necessary for a fully functional surfactant [46]. Moreover, ROS can inactivate surfactant by structural and functional modifications to surfactant proteins SP-B and SP-C [47]. Initial in vitro data suggested that the pH of lysosomes would increase as a result of CAD trapping, but subsequent, more detailed studies do not support this view [30][35][48][49]. Data in an animal model show, for example, that CQ (40 mg/kg body weight) will transiently increase hepatocyte lysosomal pH from 4.8 to 6.8 for about 2 h, followed by a return to pH 4.8 lasting for at least 10 h [49]. Maintaining an acidic lysosomal pH in the face of CQ accumulation necessitates an increased ATP consumption by V-ATPase and an increased proton flux into the lysosomal lumen.
5. Endosomal-Lysosomal Proton Fluxes, CQ/HCQ and SARS-CoV-2
A third well-studied effect caused by CADS (see Figure 2) is a generalized lysosomal accumulation of phospholipids termed phospholipidosis. Phospholipidosis is a lysosomal storage disorder characterized by an abnormal accumulation of phospholipids in the form of lamellar bodies [30]. Drugs causing phospholipidosis are recognized as being potentially toxic by the pharmaceutical industry [50][51]. The underlying mechanism(s) for phospholipidosis remains a matter of some controversy. Most likely, CAD inhibition of lysosomal lipid degradation enzymes is involved [30]. Studies in an animal model indicate that CADs can induce pulmonary lesions characterized by large foamy macrophages in the alveolar spaces [51]. The foamy alveolar macrophages show a typical CAD-induced lysosomal phospholipidosis with the potential to interfere with the phagocytosis and catabolism of pulmonary surfactant [51]. The potential pathophysiological consequences of phospholipidosis remain an area of active interest. The view that lysosomes are relatively inert organelles with a narrow degradative function is rapidly changing [36]. We now appreciate that lysosomes are dynamic organelles playing a central role in a wide variety of signaling pathways affecting immune responses, viral infectivity, the inactivation of microbes, and inflammation [35][52][53]. Alterations in luminal proton fluxes can be induced by both CQ/HCQ and ROS (see below). Proton fluxes can, in turn, influence both the levels and types of ROS [54]. Lysosomes can fuse with endosomes, thereby delivering the endocytosed cargo to lysosomes [55]. Pioneering work by Burkard et al. shows that coronavirus entry into cells can occur via this endolysosomal (or endocytic) pathway [56]. The endolysosomal pathway is under intense scrutiny due to its role as a target for COVID-19 therapy [57]. The spike glycoprotein (S) of SARS-CoV-2 is proteolytically cleaved by cellular serine protease TMPRSS2 into two subunits, S1 and S2 [58]. The S1 viral protein binds to the host cell angiotensin-converting enzyme 2 (ACE2) plasma membrane protein. The SARS-CoV-2 interaction with ACE2 initiates the formation of clathrin-coated pits, which serve as SARS-CoV-2 entry receptors. The virus is then brought into the cell’s cytoplasm via endocytosis with the formation of early endosomes. The early endosomes with SARS-CoV-2 subsequently form late endosomes that fuse with lysosomes. The S2 protein subunit promotes the fusion of the viral membrane with cellular membranes [57][58][59]. Encouragingly, sera from recovered SARS patients can block SARS-CoV-2 host cell entry in a cell culture model [58]. The notion that CQ/HCQ could alkalinize endosomal-lysosomal pH and thereby inhibit the replication of viruses requiring an acidic pH has been an attractive hypothesis [60]. This potential mechanism is often proposed as a rationale for the CQ/HCQ treatment of COVID-19 [61]. Nevertheless, the references cited above cast doubt on this hypothesis, since CQ/HCQ endosomal-lysosomal alkalinization appears to be only transient, lasting only a few hours, but long enough to confound short-term in vitro experiments.As mentioned above, CQ/HCQ are likely to increase cellular ATP consumption and increase proton flux across the lysosomal membrane. Under conditions where ATP production is decreased (e.g., mitochondrial uncoupling or hypoxia) sufficiently to inhibit V-ATPase activity, it might be possible for CQ/HCQ to induce some degree of endosomal-lysosomal alkalinization [62]. Low V-ATPase protein expression could also cause CQ/HCQ alkalinization. This avenue of research is not well studied. Intracellular ATP levels are also a key determinant governing the mode of cell death: lack of ATP favors necrosis over apoptosis [63]. Although beyond the scope of this review, it should be noted that many viruses have evolved molecular mechanisms to modulate modes of cell death to their advantage [64]. Blocking apoptosis and the subsequent killing of virally infected cells is one such mechanism
[65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][64129]
.
6. Conclusions
The potential role of ROS in COVID-19 remains to be fully elucidated. In the case of influenza A viruses, inhibition of ROS production can reduce inflammation, as measured by a decrease in virally induced cytokine production [15130]. The measurement of oxidative stress parameters in CQ/HCQ COVID-19 trials would be of clinical value. Quantification of plasma F2-isoprostanes, specifically 8-epi-prostaglandin F2α, is a clinically useful and sensitive assay for assessing in vivo lipid peroxidation and oxidative stress [65131]. Similarly, the plasma ratio of α-tocopherol quinine to α-tocopherol is an excellent indicator of antioxidant status [65132]. Marcello et al. recently found that two oxysterols, 7-ketocholesterol and 7-beta-hydroxycholesterol, were increased in the serum COVID-19 patients, particularly those with severe signs. These two oxysterols are also useful biomarkers for in vivo lipid peroxidation [66133].
1,2,3,4,5,6]. The FDA currently approves CQ/HCQ for use as antimalarials, the treatment of rheumatoid arthritis, and systemic lupus erythematosus (SLE) where they act by immunomodulatory mechanisms [7]. HCQ reduces disease activity in SLE patients but has no significant effect on pro-inflammatory cytokines [8]. A recent literature review highlights many immunomodulatory mechanisms influenced by CQ/HCQ and emphasizes our lack of current knowledge concerning their potential effects on immune responses in COVID-19 patients [9]. CQ/HCQ use may lead to unknown alterations in immune responses in COVID-19 patients, including diminished innate immune responses as well as potential modifications of the B and T cell responses to the COVID-19 virus [9].Figure 1. The organic structures of (a) hydroxychloroquine (HCQ); (b) chloroquine and; (c) heme. Chloroquine can form a membrane bound complex with heme that can promote lipid peroxidation (see Figure 4 and text).Both in vitro and in vivo animal experiments have demonstrated the anti-coronavirus activity of CQ /HCQ [10,11,12]. Moreover, CQ/HCQ are broadly available, cost-effective, and therefore attractive potential therapies for viral pandemics without an effective vaccine. A review by Cortegiani et al. on the efficacy and safety of CQ for treating COVID-19 concluded that available pre-clinical evidence was sufficiently robust to justify initiating high-quality clinical trials [4]. Based on available data, the FDA initially published an emergency use authorization of CQ/HCQ (on 28 March 2020) for COVID-19 treatment. The FDA subsequently revoked this emergency use authorization (on 15 June 2020) due to new clinical data suggesting CQ/HCQ were ineffective and raising concerns regarding “serious cardiac adverse events and other potential serious side effects” [13]. Nevertheless, the FDA states “additional clinical trials continue to evaluate the potential benefit of these drugs in treating or preventing COVID-19. “The molecular basis for the potential antiviral activity of CQ/HCQ are not fully understood, and multiple mechanisms have been proposed [9]. This review will explore the hypothesis that alterations in proton fluxes and redox physiology induced by CQ/HCQ are relevant to their potential antiviral activity and side-effects. The production of reactive oxygen species (ROS) and cellular/subcellular proton fluxes are interdependent factors modulating many antimicrobial immune responses [14]. We will also highlight concerns that CQ/HCQ-induced oxidative stress could be problematic in the treatment of critically ill COVID-19 patients. The “cytokine storm” occurring in some severe forms of viral infection (e.g., influenza A viruses) is associated with increased oxidative stress as well as increased morbidity and mortality [15]. Accumulating evidence suggests that this cytokine storm is a significant cause of ARDS and multiorgan failure in COVID-19 patients [16]. Considerable evidence suggests that parasite-specific oxidative stress induced by CQ/HCQ treatment accounts for the antimalarial activity of these drugs [17]. Less appreciated, however, is evidence (see below) showing that CQ/HCQ, by themselves, are pro-oxidants that can increase oxidative stress parameters [18,19,20,21]. Viral infections are also generally accompanied by oxidative stress with potential pathophysiological consequences [22]. As mentioned above, compelling evidence supports the view that patients with COVID-19 are at increased risk of developing ARDS and subsequent death from respiratory failure [23]. Decades of research have demonstrated the central role of oxidative stress in ARDS pathophysiology [24,25,26,27]. Understanding the potential role of oxidative stress in COVID-19 is critical, since potential anti-COVID-19 drug candidates that increase oxidative stress have the potential to exacerbate ARDS pathophysiology.
2. CQ/HCQ Pharmacology and Alterations in Subcellular Organelles Proton Fluxes
Before reviewing the role of CQ/HCQ in redox physiology and SARS-CoV-2 infection, we will briefly summarize the basics of CQ/HCQ pharmacology (see Figure 2) with an emphasis on CQ/HCQ-induced alterations in subcellular organelle proton fluxes. CQ/HCQ-induced changes in proton fluxes will have a direct effect on the type of ROS present, and thereby influence their potential effects on redox-sensitive signal transduction pathways and potential molecular and cellular damage. Among the key redox-sensitive pathways are those controlled by transcription factor nuclear factor erythroid 2 p45-related factor 2 (NRF2), hypoxia-inducible transcription factors (HIFs), nuclear factor-κB (NF-κB) transcription factors, and activator protein-1 (AP-1) transcription factors (see below as well). Excellent, comprehensive reviews of CQ/HCQ pharmacokinetics and their side-effects are available [9,28,29].Figure 2. Chloroquine (CQ) and hydroxychloroquine (HCQ) are cationic amphiphilic drugs (CADs) that share a set of common pharmacological properties: (a) they can partition into biomembranes; (b) accumulate in the lumens of acidic subcellular organelles, and; (c) induce a generalized phospholipidosis, which is the lysosomal accumulation of phospholipids.
3. CQ/HCQ Bind to Biological Membranes and Alters their Structure/Function
After many decades of research, some of the least controversial characteristics of CQ/HCQ are the ability of these weakly basic and lipophilic compounds: (1) to bind biological membranes; (2) to accumulate in the lumens of acidic subcellular vesicles and alter proton fluxes; (3) to induce generalized cellular phospholipidosis. These effects are relevant to the production of ROS, proton fluxes and the immunomodulatory and potential antiviral activities of CQ/HCQ.
CQ and HCQ both belong to a class of compounds (see Figure 1 and Figure 2) termed cationic amphiphilic drugs or CADs [30]. In plasma (pH 7.4) and cellular cytoplasm (pH 7.2), the divalent forms of CQ/HCQ are the dominant species. At pH 7.4 CQ, CQH+ (monovalent), and CQH22+ (divalent) are present at 0.026, 16.74, and 83.23%, respectively (reaction 1). Unambiguous in vitro experiments show that CQH22+ binds to the phospholipid bilayers of multilamellar liposomes with a robust partition coefficient [31]. Phosphatidylserine (PS) liposomes, with a negative charge, binds CQH22+ with a particularly high partition coefficient [31]. PS plays a central role in apoptosis. Proton NMR studies have confirmed the binding of CQH22+ to phosphatidylcholine liposomes [32]. As expected, resonance signals from protons on the hydrophobic ring carbons of CADs are affected by association with liposomes [32].
The binding of CADs to lipid bilayers is stabilized by nonspecific hydrophobic and ionic interactions [31]. Lipid bilayers are present in all cells and many subcellular organelles. It follows that CADs can accumulate, to some extent, in the cells and membranous organelles of most tissues. CAD accumulation can potentially affect many functions of all biological membranes by altering their structure and functions, e.g., membrane-bound lysosomal phosphatases and hydrolases [33].
4. CQ/HCQ Accumulate in the Lumens of Acidic Subcellular Vesicles and Alter Proton Fluxes
It is also well established that CADs accumulate in the lumens of acidic subcellular vesicles. The neutral forms of CADs can freely diffuse through the hydrophobic domain of lipid bilayers. For acidified vesicles, such as endosomes, lysosomes, M2 phagosomes, and pulmonary lysosomal lamellar bodies, the neutral forms of CADs will become charged inside the lumen and will no longer be able to diffuse out [28]. Acidified vesicles utilize a vacuolar proton-pumping ATPase (V-ATPase) to maintain a low pH by pumping protons across the vesicular membrane into the lumen [34]. For lysosomes attempting to maintain a low pH (pH 4.8) in the presence of CQ/HCQ, there will be a gradual luminal accumulation of CQ/HCQ, eventually leveling off at levels as high as 20 mM [28,35]. This process is termed “lysosomal trapping,” and trapped CADs are denoted as “lysosomotropic” [35,36,37]. CQ shows a wide variation in lysosomal trapping between organs with lungs > kidney = brain = liver > diaphragm = heart = skeletal muscles > adipose tissue [38]. The high CQ accumulation in the lungs is relevant to respiratory distress disorders [39].Lamellar bodies are a type of acidified lysosome found in type II alveolar cells (T2ACs), and keratinocytes and these organelles are known to accumulate weak bases such as CQ/HCQ [40,41]. The lamellar bodies found in T2ACs secrete surfactant, which is essential for pulmonary alveoli gas exchange [42]. T2ACs are particularly relevant to COVID-19, since these cells express angiotensin-converting enzyme-2 (ACE-2), which SARS-CoV-2 utilizes as a receptor to enter the lungs. T2AC cells are preferentially infected by SARS-CoV-2, potentially contributing to a reduced secretion or function of surfactant with a resulting loss of pulmonary compliance [43]. Reduced pulmonary compliance is a typical characteristic of ARDS, but exogenous surfactant treatment has not proven to be therapeutically effective [44,45]. Nevertheless, there is a pharmaceutical interest in testing surfactant therapy in COVID-19 patients. CQ interferes with the processing of surfactant protein C, which is necessary for a fully functional surfactant [46]. Moreover, ROS can inactivate surfactant by structural and functional modifications to surfactant proteins SP-B and SP-C [47].Initial in vitro data suggested that the pH of lysosomes would increase as a result of CAD trapping, but subsequent, more detailed studies do not support this view [30,35,48,49]. Data in an animal model show, for example, that CQ (40 mg/kg body weight) will transiently increase hepatocyte lysosomal pH from 4.8 to 6.8 for about 2 h, followed by a return to pH 4.8 lasting for at least 10 h [49]. Maintaining an acidic lysosomal pH in the face of CQ accumulation necessitates an increased ATP consumption by V-ATPase and an increased proton flux into the lysosomal lumen.
5. Endosomal-Lysosomal Proton Fluxes, CQ/HCQ and SARS-CoV-2
A third well-studied effect caused by CADS (see Figure 2) is a generalized lysosomal accumulation of phospholipids termed phospholipidosis. Phospholipidosis is a lysosomal storage disorder characterized by an abnormal accumulation of phospholipids in the form of lamellar bodies [30]. Drugs causing phospholipidosis are recognized as being potentially toxic by the pharmaceutical industry [50,51]. The underlying mechanism(s) for phospholipidosis remains a matter of some controversy. Most likely, CAD inhibition of lysosomal lipid degradation enzymes is involved [30]. Studies in an animal model indicate that CADs can induce pulmonary lesions characterized by large foamy macrophages in the alveolar spaces [51]. The foamy alveolar macrophages show a typical CAD-induced lysosomal phospholipidosis with the potential to interfere with the phagocytosis and catabolism of pulmonary surfactant [51]. The potential pathophysiological consequences of phospholipidosis remain an area of active interest. The view that lysosomes are relatively inert organelles with a narrow degradative function is rapidly changing [36]. We now appreciate that lysosomes are dynamic organelles playing a central role in a wide variety of signaling pathways affecting immune responses, viral infectivity, the inactivation of microbes, and inflammation [35,52,53]. Alterations in luminal proton fluxes can be induced by both CQ/HCQ and ROS (see below). Proton fluxes can, in turn, influence both the levels and types of ROS [54].Lysosomes can fuse with endosomes, thereby delivering the endocytosed cargo to lysosomes [55]. Pioneering work by Burkard et al. shows that coronavirus entry into cells can occur via this endolysosomal (or endocytic) pathway [56]. The endolysosomal pathway is under intense scrutiny due to its role as a target for COVID-19 therapy [57]. The spike glycoprotein (S) of SARS-CoV-2 is proteolytically cleaved by cellular serine protease TMPRSS2 into two subunits, S1 and S2 [58]. The S1 viral protein binds to the host cell angiotensin-converting enzyme 2 (ACE2) plasma membrane protein. The SARS-CoV-2 interaction with ACE2 initiates the formation of clathrin-coated pits, which serve as SARS-CoV-2 entry receptors. The virus is then brought into the cell’s cytoplasm via endocytosis with the formation of early endosomes. The early endosomes with SARS-CoV-2 subsequently form late endosomes that fuse with lysosomes. The S2 protein subunit promotes the fusion of the viral membrane with cellular membranes [57,58,59]. Encouragingly, sera from recovered SARS patients can block SARS-CoV-2 host cell entry in a cell culture model [58].The notion that CQ/HCQ could alkalinize endosomal-lysosomal pH and thereby inhibit the replication of viruses requiring an acidic pH has been an attractive hypothesis [60]. This potential mechanism is often proposed as a rationale for the CQ/HCQ treatment of COVID-19 [61]. Nevertheless, the references cited above cast doubt on this hypothesis, since CQ/HCQ endosomal-lysosomal alkalinization appears to be only transient, lasting only a few hours, but long enough to confound short-term in vitro experiments.As mentioned above, CQ/HCQ are likely to increase cellular ATP consumption and increase proton flux across the lysosomal membrane. Under conditions where ATP production is decreased (e.g., mitochondrial uncoupling or hypoxia) sufficiently to inhibit V-ATPase activity, it might be possible for CQ/HCQ to induce some degree of endosomal-lysosomal alkalinization [62]. Low V-ATPase protein expression could also cause CQ/HCQ alkalinization. This avenue of research is not well studied. Intracellular ATP levels are also a key determinant governing the mode of cell death: lack of ATP favors necrosis over apoptosis [63]. Although beyond the scope of this review, it should be noted that many viruses have evolved molecular mechanisms to modulate modes of cell death to their advantage [64]. Blocking apoptosis and the subsequent killing of virally infected cells is one such mechanism [64].
6. Conclusions
The potential role of ROS in COVID-19 remains to be fully elucidated. In the case of influenza A viruses, inhibition of ROS production can reduce inflammation, as measured by a decrease in virally induced cytokine production [15]. The measurement of oxidative stress parameters in CQ/HCQ COVID-19 trials would be of clinical value. Quantification of plasma F2-isoprostanes, specifically 8-epi-prostaglandin F2α, is a clinically useful and sensitive assay for assessing in vivo lipid peroxidation and oxidative stress [132]. Similarly, the plasma ratio of α-tocopherol quinine to α-tocopherol is an excellent indicator of antioxidant status [132]. Marcello et al. recently found that two oxysterols, 7-ketocholesterol and 7-beta-hydroxycholesterol, were increased in the serum COVID-19 patients, particularly those with severe signs. These two oxysterols are also useful biomarkers for in vivo lipid peroxidation [133].
References
- Duan, Y.J.; Liu, Q.; Zhao, S.Q.; Huang, F.; Ren, L.; Liu, L.; Zhou, Y.W. The Trial of Chloroquine in the Treatment of Corona Virus Disease 2019 (COVID-19) and Its Research Progress in Forensic Toxicology. Fa Yi Xue Za Zhi 2020, 36, doi:10.12116/j.issn.1004-5619.2020.02.001.
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 105949, doi:10.1016/j.ijantimicag.2020.105949.
- Zhou, D.; Dai, S.M.; Tong, Q. COVID-19: A recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J. Antimicrob. Chemother. 2020, doi:10.1093/jac/dkaa114.
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020, doi:10.1016/j.jcrc.2020.03.005.
- Yazdany, J.; Kim, A.H.J. Use of Hydroxychloroquine and Chloroquine During the COVID-19 Pandemic: What Every Clinician Should Know. Ann. Intern. Med. 2020, doi:10.7326/M20-1334.
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16, doi:10.1038/s41421-020-0156-0.
- Shippey, E.A.; Wagler, V.D.; Collamer, A.N. Hydroxychloroquine: An old drug with new relevance. Cleve Clin. J. Med. 2018, 85, 459–467, doi:10.3949/ccjm.85a.17034.
- Willis, R.; Seif, A.M.; McGwin, G.; Martinez-Martinez, L.A.; González, E.B.; Dang, N.; Papalardo, E.; Liu, J.; Vilá, L.M.; Reveille, J.D.; et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: Data from LUMINA (LXXV), a multiethnic US cohort. Lupus 2012, 21, 830–835, doi:10.1177/0961203312437270.
- Meyerowitz, E.A.; Vannier, A.G.L.; Friesen, M.G.N.; Schoenfeld, S.; Gelfand, J.A.; Callahan, M.V.; Kim, A.Y.; Reeves, P.M.; Poznansky, M.C. Rethinking the role of hydroxychloroquine in the treatment of COVID-19. FASEB J. 2020, 34, 6027–6037, doi:10.1096/fj.202000919.
- Keyaerts, E.; Li, S.; Vijgen, L.; Rysman, E.; Verbeeck, J.; Van Ranst, M.; Maes, P. Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice. Antimicrob. Agents Chemother. 2009, 53, 3416–3421, doi:10.1128/AAC.01509-08.
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, doi:10.1093/cid/ciaa237.
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69, doi:10.1186/1743-422X-2-69.
- FDA. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-chloroquine-and. Availabe online: (accessed on 10/17/2020).
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341, doi:10.1091/mbc.E14-05-0967.
- Ye, S.; Lowther, S.; Stambas, J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J. Virol. 2015, 89, 2672–2683, doi:10.1128/JVI.03529-14.
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613, doi:10.1016/j.jinf.2020.03.037.
- Herraiz, T.; Guillén, H.; González-Peña, D.; Arán, V.J. Antimalarial Quinoline Drugs Inhibit β-Hematin and Increase Free Hemin Catalyzing Peroxidative Reactions and Inhibition of Cysteine Proteases. Sci. Rep. 2019, 9, 15398, doi:10.1038/s41598-019-51604-z.
- Toler, S.M.; Noe, D.; Sharma, A. Selective enhancement of cellular oxidative stress by chloroquine: Implications for the treatment of glioblastoma multiforme. Neurosurg. Focus 2006, 21, E10, doi:10.3171/foc.2006.21.6.1.
- Farombi, E.O.; Shyntum, Y.Y.; Emerole, G.O. Influence of chloroquine treatment and Plasmodium falciparum malaria infection on some enzymatic and non-enzymatic antioxidant defense indices in humans. Drug Chem. Toxicol. 2003, 26, 59–71, doi:10.1081/dct-120017558.
- Farombi, E.O. Genotoxicity of chloroquine in rat liver cells: Protective role of free radical scavengers. Cell Biol. Toxicol. 2006, 22, 159–167, doi:10.1007/s10565-006-0173-2.
- Lehane, A.M.; McDevitt, C.A.; Kirk, K.; Fidock, D.A. Degrees of chloroquine resistance in Plasmodium - is the redox system involved? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 47–57, doi:10.1016/j.ijpddr.2011.11.001.
- Camini, F.C.; da Silva Caetano, C.C.; Almeida, L.T.; de Brito Magalhães, C.L. Implications of oxidative stress on viral pathogenesis. Arch. Virol. 2017, 162, 907–917, doi:10.1007/s00705-016-3187-y.
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Du, C.; Zhang, Y.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, doi:10.1001/jamainternmed.2020.0994.
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431, doi:10.1165/rcmb.F278.
- Rocksén, D.; Ekstrand-Hammarström, B.; Johansson, L.; Bucht, A. Vitamin E reduces transendothelial migration of neutrophils and prevents lung injury in endotoxin-induced airway inflammation. Am. J. Respir. Cell Mol. Biol. 2003, 28, 199–207, doi:10.1165/rcmb.4899.
- Stone, W.L.; Mukherjee, S.; Smith, M.; Das, S.K. Therapeutic uses of antioxidant liposomes. Methods Molecular Biol. (Clifton N. J.) 2002, 199, 145–161, doi:10.1385/1-59259-175-2:145.
- Stone, W.L.; Smith, M. Therapeutic uses of antioxidant liposomes. Mol. Biotechnol. 2004, 27, 217–230, doi:10.1385/MB:27:3:217.
- Al-Bari, M.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621, doi:10.1093/jac/dkv018.
- Schrezenmeier, E.; Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166, doi:10.1038/s41584-020-0372-x.
- Breiden, B.; Sandhoff, K. Emerging mechanisms of drug-induced phospholipidosis. Biol. Chem. 2019, 401, 31–46, doi:10.1515/hsz-2019-0270.
- Lüllmann, H.; Wehling, M. The binding of drugs to different polar lipids in vitro. Biochem. Pharmacol. 1979, 28, 3409–3415, doi:10.1016/0006-2952(79)90080-7.
- Seydel, J.K.; Wassermann, O. NMR-studies on the molecular basis of drug-induced phospholipidosis--II. Interaction between several amphiphilic drugs and phospholipids. Biochem. Pharmacol. 1976, 25, 2357–2364, doi:10.1016/0006-2952(76)90028-9.
- Halliwell, W.H. Cationic amphiphilic drug-induced phospholipidosis. Toxicol. Pathol. 1997, 25, 53–60, doi:10.1177/019262339702500111.
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929, doi:10.1038/nrm2272.
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12, e0173771, doi:10.1371/journal.pone.0173771.
- Homewood, C.A.; Warhurst, D.C.; Peters, W.; Baggaley, V.C. Lysosomes, pH and the anti-malarial action of chloroquine. Nature 1972, 235, 50–52, doi:10.1038/235050a0.
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331, doi:10.1073/pnas.75.7.3327.
- Daniel, W.A.; Bickel, M.H.; Honegger, U.E. The contribution of lysosomal trapping in the uptake of desipramine and chloroquine by different tissues. Pharmacol. Toxicol. 1995, 77, 402–406, doi:10.1111/j.1600-0773.1995.tb01050.x.
- Ndiaye, N.; Petrognani, R.; Diatta, B.; Seck, M.; Theobald, X.; Adnet, P. [Chloroquine poisoning with respiratory distress and fatal outcome]. Ann. Fr. Anesth. Reanim. 1999, 18, 683–685, doi:10.1016/s0750-7658(99)80157-9.
- Fois, G.; Hobi, N.; Felder, E.; Ziegler, A.; Miklavc, P.; Walther, P.; Radermacher, P.; Haller, T.; Dietl, P. A new role for an old drug: Ambroxol triggers lysosomal exocytosis via pH-dependent Ca²⁺ release from acidic Ca²⁺ stores. Cell Calcium 2015, 58, 628–637, doi:10.1016/j.ceca.2015.10.002.
- Schmitz, G.; Müller, G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J. Lipid Res. 1991, 32, 1539–1570.
- Akella, A.; Deshpande, S.B. Pulmonary surfactants and their role in pathophysiology of lung disorders. Indian J. Exp. Biol 2013, 51, 5–22.
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55, doi:10.1183/13993003.00607-2020.
- Zhang, L.N.; Sun, J.P.; Xue, X.Y.; Wang, J.X. Exogenous pulmonary surfactant for acute respiratory distress syndrome in adults: A systematic review and meta-analysis. Exp. Ther. Med. 2013, 5, 237–242, doi:10.3892/etm.2012.746.
- Meng, S.S.; Chang, W.; Lu, Z.H.; Xie, J.F.; Qiu, H.B.; Yang, Y.; Guo, F.M. Effect of surfactant administration on outcomes of adult patients in acute respiratory distress syndrome: A meta-analysis of randomized controlled trials. BMC Pulm. Med. 2019, 19, 9, doi:10.1186/s12890-018-0761-y.
- Beers, M.F. Inhibition of cellular processing of surfactant protein C by drugs affecting intracellular pH gradients. J. Biol. Chem. 1996, 271, 14361–14370, doi:10.1074/jbc.271.24.14361.
- Rodríguez-Capote, K.; Manzanares, D.; Haines, T.; Possmayer, F. Reactive oxygen species inactivation of surfactant involves structural and functional alterations to surfactant proteins SP-B and SP-C. Biophys. J. 2006, 90, 2808–2821, doi:10.1529/biophysj.105.073106.
- Hamaguchi, R.; Haginaka, J.; Tanimoto, T.; Kuroda, Y. Maintenance of luminal pH and protease activity in lysosomes/late endosomes by vacuolar ATPase in chlorpromazine-treated RAW264 cells accumulating phospholipids. Cell Biol. Toxicol. 2014, 30, 67–77, doi:10.1007/s10565-014-9269-2.
- Tietz, P.S.; Yamazaki, K.; LaRusso, N.F. Time-dependent effects of chloroquine on pH of hepatocyte lysosomes. Biochem. Pharmacol. 1990, 40, 1419–1421, doi:10.1016/0006-2952(90)90414-g.
- Shayman, J.A.; Abe, A. Drug induced phospholipidosis: An acquired lysosomal storage disorder. Biochim. Biophys. Acta 2013, 1831, 602–611, doi:10.1016/j.bbalip.2012.08.013.
- Robison, R.L.; Visscher, G.E.; Roberts, S.A.; Engstrom, R.G.; Hartman, H.A.; Ballard, F.H. Generalized phospholipidosis induced by an amphiphilic cationic psychotropic drug. Toxicol. Pathol. 1985, 13, 335–348, doi:10.1177/019262338501300410.
- Jankowski, A.; Scott, C.C.; Grinstein, S. Determinants of the phagosomal pH in neutrophils. J. Biol. Chem. 2002, 277, 6059–6066, doi:10.1074/jbc.M110059200.
- Jankowski, A.; Grinstein, S. Modulation of the cytosolic and phagosomal pH by the NADPH oxidase. Antioxid. Redox Signal. 2002, 4, 61–68, doi:10.1089/152308602753625861.
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic 2013, 14, 1118–1131, doi:10.1111/tra.12115.
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632, doi:10.1038/nrm2217.
- Burkard, C.; Verheije, M.H.; Wicht, O.; van Kasteren, S.I.; van Kuppeveld, F.J.; Haagmans, B.L.; Pelkmans, L.; Rottier, P.J.; Bosch, B.J.; de Haan, C.A. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014, 10, e1004502, doi:10.1371/journal.ppat.1004502.
- Yang, N.; Shen, H.M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731, doi:10.7150/ijbs.45498.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278, doi:10.1016/j.cell.2020.02.052.
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729, doi:10.1128/JVI.00253-07.
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5, e00293, doi:10.1002/prp2.293.
- Singh, A.K.; Singh, A.; Shaikh, A.; Singh, R.; Misra, A. Chloroquine and hydroxychloroquine in the treatment of COVID-19 with or without diabetes: A systematic search and a narrative review with a special reference to India and other developing countries. Diabetes Metab. Syndr. 2020, 14, 241–246, doi:10.1016/j.dsx.2020.03.011.
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795, doi:10.3390/cells8080795.
- Tsujimoto, Y. Apoptosis and necrosis: Intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997, 4, 429–434, doi:10.1038/sj.cdd.4400262.
- Kvansakul, M. Viral Infection and Apoptosis. Viruses 2017, 9, 356, oi:10.3390/v9120356.
- Stone, W.L.; LeClair, I.; Ponder, T.; Baggs, G.; Reis, B.B. Infants discriminate between natural and synthetic vitamin E. Am. J. Clin. Nutr. 2003, 77, 899–906.Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619, doi:10.1002/eji.200940168.
- Marcello, A.; Civra, A.; Milan Bonotto, R.; Nascimento Alves, L.; Rajasekharan, S.; Giacobone, C.; Caccia, C.; Cavalli, R.; Adami, M.; Brambilla, P.; et al. The cholesterol metabolite 27-hydroxycholesterol inhibits SARS-CoV-2 and is markedly decreased in COVID-19 patients. Redox Biol. 2020, 36, 101682, doi:10.1016/j.redox.2020.101682.Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407, doi:10.3390/ijms20102407.
- Stone, W.L.; Basit, H.; Mohiuddin, S.S. Biochemistry, Antioxidants. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2019.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13, doi:10.1042/BJ20081386.
- Onukwufor, J.O.; Berry, B.J.; Wojtovich, A.P. Physiologic Implications of Reactive Oxygen Species Production by Mitochondrial Complex I Reverse Electron Transport. Antioxidants (Basel) 2019, 8, doi:10.3390/antiox8080285.
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513, doi:10.1016/j.tibs.2010.04.002.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, doi:10.1038/sigtrans.2017.23.
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115, doi:10.1038/cr.2010.178.
- Park, J.; Kwon, D.; Choi, C.; Oh, J.W.; Benveniste, E.N. Chloroquine induces activation of nuclear factor-kappaB and subsequent expression of pro-inflammatory cytokines by human astroglial cells. J. Neurochem. 2003, 84, 1266–1274, doi:10.1046/j.1471-4159.2003.01623.x.
- Yang, S.; Qiang, L.; Sample, A.; Shah, P.; He, Y.Y. NF-κB Signaling Activation Induced by Chloroquine Requires Autophagosome, p62 Protein, and c-Jun N-terminal Kinase (JNK) Signaling and Promotes Tumor Cell Resistance. J. Biol. Chem. 2017, 292, 3379–3388, doi:10.1074/jbc.M116.756536.
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656, doi:10.1146/annurev.pharmtox.47.120505.105110.
- Anderson, E.J.; Katunga, L.A.; Willis, M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012, 39, 179–193, doi:10.1111/j.1440-1681.2011.05641.x.
- Wilkerson, R.G.; Adler, J.D.; Shah, N.G.; Brown, R. Silent hypoxia: A harbinger of clinical deterioration in patients with COVID-19. Am. J. Emerg. Med. 2020, doi:10.1016/j.ajem.2020.05.044.
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell Longev. 2014, 2014, 360438, doi:10.1155/2014/360438.
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921, doi:10.1074/jbc.M900392200.
- Deepalakshmi, P.D.; Parasakthy, K.; Shanthi, S.; Devaraj, N.S. Effect of chloroquine on rat liver mitochondria. Indian J. Exp. Biol. 1994, 32, 797–799.
- Katewa, S.D.; Katyare, S.S. Treatment with antimalarials adversely affects the oxidative energy metabolism in rat liver mitochondria. Drug Chem. Toxicol. 2004, 27, 41–53, doi:10.1081/dct-120027898.
- Berry, B.J.; Trewin, A.J.; Amitrano, A.M.; Kim, M.; Wojtovich, A.P. Use the Protonmotive Force: Mitochondrial Uncoupling and Reactive Oxygen Species. J. Mol. Biol. 2018, 430, 3873–3891, doi:10.1016/j.jmb.2018.03.025.
- Nainu, F.; Shiratsuchi, A.; Nakanishi, Y. Induction of Apoptosis and Subsequent Phagocytosis of Virus-Infected Cells As an Antiviral Mechanism. Front. Immunol. 2017, 8, 1220, doi:10.3389/fimmu.2017.01220.
- Mills, E.L.; Debets-Ossenkopp, Y.; Verbrugh, H.A.; Verhoef, J. Initiation of the respiratory burst of human neutrophils by influenza virus. Infect. Immun. 1981, 32, 1200–1205.
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32, doi:10.1016/j.cytogfr.2020.05.003.
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, doi:10.1016/S0140-6736(20)30937-5.
- Westlin, W.F.; Gimbrone, M.A. Neutrophil-mediated damage to human vascular endothelium. Role of cytokine activation. Am. J. Pathol. 1993, 142, 117–128.
- Szmitko, P.E.; Wang, C.H.; Weisel, R.D.; de Almeida, J.R.; Anderson, T.J.; Verma, S. New markers of inflammation and endothelial cell activation: Part I. Circulation 2003, 108, 1917–1923, doi:10.1161/01.CIR.0000089190.95415.9F.
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660, doi:10.1089/ars.2012.4827.
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016, 130, 479–490, doi:10.1042/CS20150660.
- Zemans, R.L.; Matthay, M.A. What drives neutrophils to the alveoli in ARDS? Thorax 2017, 72, 1–3, doi:10.1136/thoraxjnl-2016-209170.
- Huang, X.; Xiu, H.; Zhang, S.; Zhang, G. The Role of Macrophages in the Pathogenesis of ALI/ARDS. Med. Inflamm. 2018, 2018, 1264913, doi:10.1155/2018/1264913.
- Minari, J.B.; Oloyede, O.B. Immunosupressive effect of chloroquine through the inhibition of myeloperoxidase. In Proceedings of the 2nd International Conference on Clinical & Cellular Immunology, Hampton Inn Tropicana, Las Vegas, NV, USA, 15–17 October 2013.
- Labro, M.T.; Babin-Chevaye, C. Effects of amodiaquine, chloroquine, and mefloquine on human polymorphonuclear neutrophil function in vitro. Antimicrob. Agents Chemother. 1988, 32, 1124–1130, doi:10.1128/aac.32.8.1124.
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Publisher Correction: Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 1808, doi:10.1038/s41467-018-04169-w.
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193, doi:10.4049/jimmunol.1701135.
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553, doi:10.3389/fimmu.2020.00553.
- Delgado-Roche, L.; Mesta, F. Oxidative Stress as Key Player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection. Arch. Med. Res. 2020, 51, 384–387, doi:10.1016/j.arcmed.2020.04.019.
- Sugioka, Y.; Suzuki, M.; Sugioka, K.; Nakano, M. A ferriprotoporphyrin IX-chloroquine complex promotes membrane phospholipid peroxidation. A possible mechanism for antimalarial action. FEBS Lett. 1987, 223, 251–254, doi:10.1016/0014-5793(87)80299-5.
- Bhattacharyya, B.; Chatterjee, T.K.; Ghosh, J.J. Effects of chloroquine on lysosomal enzymes, NADPH-induced lipid peroxidation, and antioxidant enzymes of rat retina. Biochem. Pharmacol. 1983, 32, 2965–2968, doi:10.1016/0006-2952(83)90403-3.
- Yusuf, I.H.; Sharma, S.; Luqmani, R.; Downes, S.M. Hydroxychloroquine retinopathy. Eye 2017, 31, 828–845, doi:10.1038/eye.2016.298.
- Ogunbayo, O.A.; Adisa, R.A.; Ademowo, O.G.; Olorunsogo, O. Incidence of Chloroquine Induced Oxidative Stress in the Blood of Rabbit. Int. J. Pharmacol. 2006, 2, 121–125, doi:10.3923/ijp.2006.121.125
- Giovanella, F.; Ferreira, G.K.; de Prá, S.D.; Carvalho-Silva, M.; Gomes, L.M.; Scaini, G.; Gonçalves, R.C.; Michels, M.; Galant, L.S.; Longaretti, L.M.; et al. Effects of primaquine and chloroquine on oxidative stress parameters in rats. An. Acad. Bras. Cienc. 2015, 87, 1487–1496, doi:10.1590/0001-3765201520140637.
- Fang, L.; Neutzner, A.; Turtschi, S.; Flammer, J.; Mozaffarieh, M. Comet assay as an indirect measure of systemic oxidative stress. J. Vis. Exp. 2015, e52763, doi:10.3791/52763.
- Stone, W.L.; Farnsworth, C.C.; Dratz, E.A. A reinvestigation of the fatty acid content of bovine, rat and frog retinal rod outer segments. Exp. Eye Res. 1979, 28, 387–397, doi:0014-4835(79)90114-3 [pii].
- Song, J.H.; Fujimoto, K.; Miyazawa, T. Polyunsaturated (n-3) Fatty Acids Susceptible to Peroxidation Are Increased in Plasma and Tissue Lipids of Rats Fed Docosahexaenoic Acid–Containing Oils. J. Nutr. 2000, 130, 3028–3033, doi:10.1093/jn/130.12.3028.
- Jančinová, V.; Pažoureková, S.; Lucová, M.; Perečko, T.; Mihalová, D.; Bauerová, K.; Nosáľ, R.; Drábiková, K. Selective inhibition of extracellular oxidants liberated from human neutrophils—A new mechanism potentially involved in the anti-inflammatory activity of hydroxychloroquine. Int. Immunopharmacol. 2015, 28, 175–181, doi:10.1016/j.intimp.2015.05.048.
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71, doi:10.1126/scitranslmed.3001118.
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61, doi:10.3389/fphar.2014.00061.
- de Dios, A.C.; Tycko, R.; Ursos, L.M.B.; Roepe, P.D. NMR Studies of Chloroquine−Ferriprotoporphyrin IX Complex. J. Phys. Chem. A 2003, 107, 5821–5825, doi:10.1021/jp0342982.
- Atamna, H. Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev. 2004, 3, 303–318, doi:10.1016/j.arr.2004.02.002.
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal. 2010, 12, 233–248, doi:10.1089/ars.2009.2822.
- Roumenina, L.T.; Rayes, J.; Lacroix-Desmazes, S.; Dimitrov, J.D. Heme: Modulator of Plasma Systems in Hemolytic Diseases. Trends Mol Med 2016, 22, 200-213, doi:10.1016/j.molmed.2016.01.004.
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Wang, S.; Jonas, W.; Kirchhofer, D.; Gailani, D.; Gruber, A.; Kasthuri, R.; Key, N.S.; Mackman, N.; et al. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica 2015, 100, 308–314, doi:10.3324/haematol.2014.114728.
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040, doi:10.1182/blood.2020006000.
- Kander, T. Coagulation disorder in COVID-19. Lancet Haematol. 2020, doi:10.1016/S2352-3026(20)30218-0.
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002, 33, 457–463, doi:10.1016/s0891-5849(02)00842-0.
- Hooper, P.L. COVID-19 and heme oxygenase: Novel insight into the disease and potential therapies. Cell Stress Chaperones 2020, doi:10.1007/s12192-020-01126-9.
- Kwon, K.J.; Kim, J.N.; Kim, K.M.; Lee, J.; Ignarro, L.J.; Kim, H.-J.; Shin, C.Y.; Han, S.H. Melatonin synergistically increases resveratrol-induced heme oxygenase-1 expression through the inhibition of ubiquitin-dependent proteasome pathway: A possible role in neuroprotection. J. Pineal Res. 2011, 50, 110–123, doi:10.1111/j.1600-079X.2010.00820.x.
- Wessels, I.; Rolles, B.; Rink, L. The Potential Impact of Zinc Supplementation on COVID-19 Pathogenesis. Front. Immunol. 2020, 11, 1712.
- Velthius, A.J.W.T.; van den Worm, S.H.E.; Sims, A.C.; Baric, R.S.; Snijder, E.J.; van Hemert, M.J. Zn2+ Inhibits Coronavirus and Arterivirus RNA Polymerase Activity In Vitro and Zinc Ionophores Block the Replication of These Viruses in Cell Culture. PLOS Pathog. 2010, 6, e1001176, doi:10.1371/journal.ppat.1001176.
- Xue, J.; Moyer, A.; Peng, B.; Wu, J.; Hannafon, B.N.; Ding, W.-Q. Chloroquine Is a Zinc Ionophore. PLoS ONE 2014, 9, e109180, doi:10.1371/journal.pone.0109180.
- Wong-ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236, doi:10.1529/biophysj.107.112565.
- Dabbagh-Bazarbachi, H.; Clergeaud, G.; Quesada, I.; Ortiz, M.; O’Sullivan, C.; Fernandez-Larrea, J. Zinc Ionophore Activity of Quercetin and Epigallocatechin-gallate: From Hepa 1-6 Cells to a Liposome Model. J. Agric. Food Chem. 2014, 62, 8085–8093, doi:10.1021/jf5014633.
- NIH. https://clinicaltrials.gov/ct2/show/NCT04370782. Availabe online: (accessed on 09/17/2020).
- Yarosz, E.L.; Chang, C.H. The Role of Reactive Oxygen Species in Regulating T Cell-mediated Immunity and Disease. Immune Netw. 2018, 18, e14, doi:10.4110/in.2018.18.e14.
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271, doi:10.1038/s41422-020-0282-0.
- Hoffmann, M.; Mösbauer, K.; Hofmann-Winkler, H.; Kaul, A.; Kleine-Weber, H.; Krüger, N.; Gassen, N.C.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 2020, doi:10.1038/s41586-020-2575-3.
- Skipper, C.P.; Pastick, K.A.; Engen, N.W.; Bangdiwala, A.S.; Abassi, M.; Lofgren, S.M.; Williams, D.A.; Okafor, E.C.; Pullen, M.F.; Nicol, M.R.; et al. Hydroxychloroquine in Nonhospitalized Adults With Early COVID-19: A Randomized Trial. Ann. Intern. Med. 2020, doi:10.7326/M20-4207.
- Boulware, D.R.; Pullen, M.F.; Bangdiwala, A.S.; Pastick, K.A.; Lofgren, S.M.; Okafor, E.C.; Skipper, C.P.; Nascene, A.A.; Nicol, M.R.; Abassi, M.; et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N. Eng. J. Med. 2020, doi:10.1056/NEJMoa2016638.
- Cavalcanti, A.B.; Zampieri, F.G.; Rosa, R.G.; Azevedo, L.C.P.; Veiga, V.C.; Avezum, A.; Damiani, L.P.; Marcadenti, A.; Kawano-Dourado, L.; Lisboa, T.; et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. N. Eng. J. Med. 2020, doi:10.1056/NEJMoa2019014.
- Stone, W.L.; LeClair, I.; Ponder, T.; Baggs, G.; Reis, B.B. Infants discriminate between natural and synthetic vitamin E. Am. J. Clin. Nutr. 2003, 77, 899–906.
- Marcello, A.; Civra, A.; Milan Bonotto, R.; Nascimento Alves, L.; Rajasekharan, S.; Giacobone, C.; Caccia, C.; Cavalli, R.; Adami, M.; Brambilla, P.; et al. The cholesterol metabolite 27-hydroxycholesterol inhibits SARS-CoV-2 and is markedly decreased in COVID-19 patients. Redox Biol. 2020, 36, 101682, doi:10.1016/j.redox.2020.101682.