Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sara Napoli and Version 2 by Rita Xu.
Enhancer RNAs (eRNAs) are non-coding RNAs (ncRNAs) transcribed in enhancer regions. They play an important role in transcriptional regulation, mainly during cellular differentiation. eRNAs are tightly tissue- and cell-type specific and are induced by specific stimuli, activating promoters of target genes in turn. eRNAs usually have a very short half-life but in some cases, once activated, they can be stably expressed and acquire additional functions. Due to their critical role, eRNAs are often dysregulated in cancer and growing number of interactions with chromatin modifiers, transcription factors, and splicing machinery have been described.
- enhancer
- ncRNA
- eRNA
- transcriptional regulation
- cancer
1. Introduction
Huge progress made in understanding the human genome has come hand in hand with the discovery of its complexity. Only 1–2% of the human transcriptome codes for proteins [1]. The remaining transcriptome is represented by an overwhelming amount of non-coding RNAs (ncRNAs): ribosomal-RNA (rRNA), transfer-RNA (tRNA), small nucleolar RNA (snoRNA), micro-RNA (miRNA), but also long-noncoding-RNA (lncRNA), including competing-endogenous-RNA (ceRNA), and many more [1]. Furthermore, the genome comprises extended regulatory regions such as enhancers, defined as cis-regulatory elements spatially and temporally cooperating with promoters, able to bind transcription factors, co-regulators, and RNA polymerase II (RNApol II) to mediate target gene transcription [2]. The study of the formation of the enhancer–promoter loop crucial for the transcription initiation process showed that enhancers exert their function in an orientation independent manner. In addition, enhancers are positioned at various distances upstream or downstream the transcription starting side (TSS) of the associated gene, and they can work together in networks comprising other enhancers to control gene expression [3]. Currently, the knowledge of such complex enhancer interplay is limited mainly to enhancers organized in super enhancer (SE) regions. SEs are large clusters of enhancers (>20 kb in size), forming specific 3D structures associated with massive transcription rates and play an essential role in cell growth and differentiation, but also in disease initiation and progression of cancers [4].
The epigenetic state of enhancers is essential for their function and, indeed, it is often exploited to identify enhancers themselves and to define their genomic localization. Nucleosomes surrounding active enhancers harbor high histone H3 lysine 4 mono-methylation (H3K4me1) and histone H3 lysine 27 acetylation (H3K27ac) levels [5]. However, additional factors need to be considered for robust enhancer identification. The discovery of the binding of RNApol II to active enhancers revealed the transcription of a new subclass of ncRNAs, the enhancer RNAs (eRNAs). The latter can be used as an additional indicator for active enhancer regions beside the classical histone marks previously described [6]. Moreover, the discovery of eRNA expression added another layer of complexity to the human transcriptome, leading to intense research on the features and potential functions of this new class of ncRNAs [7].
Although more and more commonly identified and recognized, the biological roles of eRNAs are still under investigation. In the last couple of years, a growing number of studies showed that eRNAs participate in the regulation process of gene transcription [7]. First, they contribute to the enhancer–promoter loop, recruiting cohesin and mediator proteins [8][9]. They trap transcription factors, as YY1, to increase their local concentration at DNA at the site of transcription [10]. Furthermore, eRNAs are also directly involved in transcription, acting as decoys for negative elongation factor (NELF) and releasing paused RNApol II to promote the elongation process [11] (
Although more and more commonly identified and recognized, the biological roles of eRNAs are still under investigation. In the last couple of years, a growing number of studies showed that eRNAs participate in the regulation process of gene transcription [7]. First, they contribute to the enhancer–promoter loop, recruiting cohesin and mediator proteins [8,9]. They trap transcription factors, as YY1, to increase their local concentration at DNA at the site of transcription [10]. Furthermore, eRNAs are also directly involved in transcription, acting as decoys for negative elongation factor (NELF) and releasing paused RNApol II to promote the elongation process [11] (
Figure 1
).
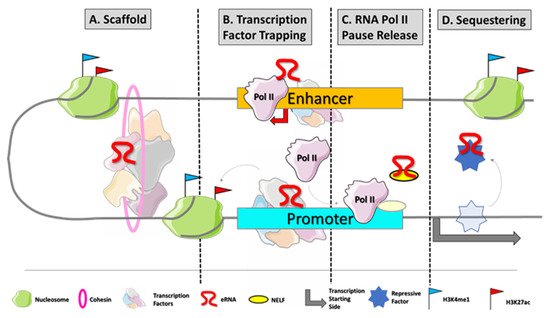
Figure 1.
eRNA biogenesis and frequently described modes of action. Enhancers are typically disengaged of nucleosomes which makes their DNA sequences easily accessible for transcription factor (TF) binding. Master TFs occupy enhancer regions at specific DNA motifs to initiate the transcriptional process by recruiting RNApol II at enhancers. Those transcripts mainly promote gene expression by different mechanisms of action: (
B
C
D
) eRNAs can act as decoys for repressive co-factors of transcription which would bind at the respective target genes.
Notably, eRNAs seem to play a role not only in homeostasis and cell development, but also in disease development and differentiation-state. They can modulate the expression of oncogenes, tumor suppressor genes, and inflammatory or cancer signaling pathways by modifying gene transcription and protein–RNA interactions [12]. The eRNAs also represent potential biomarkers of treatment response [12][13][14].
Notably, eRNAs seem to play a role not only in homeostasis and cell development, but also in disease development and differentiation-state. They can modulate the expression of oncogenes, tumor suppressor genes, and inflammatory or cancer signaling pathways by modifying gene transcription and protein–RNA interactions [12]. The eRNAs also represent potential biomarkers of treatment response [12,13,14].
2. eRNAs Instability and Methodology of Detection
eRNAs are noncoding molecules bidirectionally transcribed from enhancers by RNApol II [15]. The eRNAs are 5′-capped but less stable and shorter than mRNAs because they are retained in the nucleus and exposed to exosome-mediated decay [16]. Indeed, they are mainly unspliced and not polyadenylated. The lack of polyA causes the recruitment of exosome, because the small distance between polyA signal and transcription start site prevents the assembly of polyadenylation machinery at RNApol II C-terminal domain (CTD) [17]. eRNAs are densely located on the chromatin where they help the stabilization of enhancer–promoter (E–P) interactions [17]. These features make them rarely detectable by conventional polyA RNA-seq. Their identification and quantification is feasible by techniques such as ribosomal RNA (rRNA)-depleted total RNA-seq or cap analysis of gene expression (CAGE) [18][19][20]. Other techniques such as the global run-on sequencing (GRO-seq) [21] and its variant precision nuclear run-on sequencing (PRO-seq) [22] investigate RNApol II activity, sequencing the nascent RNA molecules, and thus, they allow the detection of even unstable transcripts. Transient transcriptome sequencing (TT-seq) is often applied to study eRNAs [23][24][25]. It is a sensitive approach to detect short-lived transcripts, which combines a short 4-thiouridine (4sU) RNA labeling pulse with an RNA fragmentation step to enrich for newly synthesized RNA. An adapted CAGE protocol, NET-CAGE, was recently developed to study TSSs of native elongating transcripts (NETs) in a strand-specific manner [26]. It sensitively identifies and quantifies true transcriptional activities of enhancers at high nucleotide resolution and allows the study of RNA synthesis and degradation rates at the TSS level. Other techniques can help in characterizing eRNA functions, linking the eRNA with its target gene. For instance, an advanced imaging protocol for single molecule fluorescence in situ hybridization (smFISH) was implemented to detect short and lowly expressed transcripts at single cell level [27]. This method uses single biotinylated short (20 nt each) probes, which, post hybridization, allow the binding of multiple copies of fluorophore per probe. The system can be multiplexed to achieve simultaneous detection of both eRNA and the induced nascent gene transcript in the same nucleus to prove their co-localization on the chromatin. Few technologies are emerging to agnostically infer eRNA function by assessing genome-wide RNA chromatin interactions. RADICL-Seq captures proximal RNA-chromatin interactions in a genome-wide manner [28]. Thanks to the enhancer–promoter looping, this technique can capture the spatial proximity of the nascent RNA with the enhancer region. This improved method overcomes limitations of similar pre-existing techniques such as mapping RNA–genome interactions (MARGI) [29], chromatin-associated RNA-sequencing (CHAR-seq) [30], and global RNA interaction with DNA by deep-sequencing (GRID-seq) [31].3. eRNAs in Development and Differentiation
The expression of eRNAs is developmental stage and cell-type specific [32][33]. eRNAs transcribed during cellular differentiation are correlated to their target gene expression [2][34] and their production explains the interaction between enhancers and transcription initiation and their proximity to promoters. Enhancer transcription is the most common rapid transcriptional adjustment occurring when cells undergo the first steps toward a state change [19]. Once the target promoter is activated, enhancer activity is no longer required, and so, after a rapid burst of production and activity, eRNAs frequently return to baseline expression levels. In some specific cases, enhancers are rapidly activated and then continuously expressed and these eRNAs might have additional functional roles still unknown [19]. Accordingly, with their pivotal function, eRNAs are involved in determining the developmental stage of cells, often cooperating to regulate the expression of their target genes [2]. General insights into the order of events that establish active enhancers have been recently provided by the application of a genome-wide multi-omics approach to study the C/EBPα-induced trans-differentiation of human precursor leukemia B cells to macrophage-like cells [25]. In particular, TT-seq allowed the identification of transcriptionally active enhancers, then paired with their putative target promoters, and the analysis of changes in transcription activity of enhancers and promoters over time [25]. Most enhancers drive the expression of a common target gene acting in an additive manner, but some enhancers can cooperate synergistically at specific loci to drive target gene transcription [25]. The interacting network of molecules that regulates differentiation is complex and involves different types of lncRNAs. A genome-wide study combined RNA sequencing (RNA-seq) and RNA reverse transcription-associated capture sequencing (RAT-seq) to map interacting targets for lncRNAs [35]. The lcnRNA Peblr20 was identified as a demanding element that helps in maintaining the pluripotent status of induced pluripotent stem cells. Peblr20 knockdown in induced pluripotent stem cells withdrew pluripotency state and, vice versa, overexpression of Peblr20 activated stemness-like genes such as Pou5F1, SOX2, and NANOG in fibroblasts, leading to improved pluripotent reprogramming. Notably, lncRNA Peblr20 recruited TET2 to the Pou5F1 enhancer site, leading to DNA demethylation at the Pou5F1 enhancer and, therefore, to Pou5F1 eRNA expression. Pou5F1 eRNA expression itself seemed to modulate the enhancer promoter looping, activating Pou5F1 expression and induced pluripotent state. Thus, Peblr20 utilizes a novel trans epigenetic eRNA activation mechanism to control the stem cell fate mediated by another ncRNA subgroup, suggesting a widely unexplored interplay between other noncoding regulatory RNAs and eRNAs [35].4. eRNAs Control Transcription by Several Layers of Sequence Specific Mechanisms
Recently, the abundance of m6A on eRNAs in correspondence of RRACH motif was demonstrated, suggesting an m6A deposition similar to that on mRNAs [36]. Lee at al. showed that the m6A reader YTHDC1 catalyzes the binding of BRD4 to m6A eRNA. In a positive feedback loop, YTHDC1 directly activates enhancers and induces eRNA transcription [37]. However, no evidence of a relationship between m6A and eRNA stability is proven, but on the other hand m6A eRNA bound by YTHDC1 helps chromatin condensation, cross talking to other coactivators and impacting enhancer activity [37]. The act of enhancer transcription generates eRNAs, and the length of these transcripts may influence their m6A levels, consequently facilitating transcriptional activation [37]. Enhancers can be active, primed, or poised, depending on different epigenetic states: active enhancers, which are H3K27ac and H3K4me1 positive and bound by the histone acetyltransferase P300, are strong transcription activators, while, when primed, they have lower H3K27ac and only drive basal transcriptional activation. On the contrary, poised enhancers present H3K4me1 and P300, but they also have the repressive histone mark H3K27me3, associated with Polycomb Repressive Complex 2 (PRC2) silencing [38]. Expression of eRNAs can be activated in poised enhancers by sequence specific mechanism driven by lncRNAs, forming triple-helix structures with DNA [39]. Triplex-based recruitment of chromatin-modifying complexes may represent a common targeting mechanism for enhancer activation. Blank-Giwojna et al. demonstrated that the antisense RNA KHPS1 forms a RNA–DNA triplex at the SPHK1 enhancer, with consequent recruitment of E2F1 and p300 [39]. The enhancer activation induces transcription of an eRNA that is required for SPHK1 expression and cell proliferation. Genomic deletion of the triplex-forming region (TFR) or prevention of KHPS1 binding to DNA impairs cell proliferation and viability [39]. The functional role of the sequence specific lncRNA–DNA binding is proven by the fact that changes in the lncRNA sequence, driving it to distinct genomic location, activating different enhancers and the expression of different genes [39]. The mechanism that leads to promoter activation involves the eRNA-mediated eviction of CTCF, which insulates eSPHK1 from the SPHK1-C promoter [39]. Numerous studies have shown that CTCF regulates enhancer–promoter interactions and the human genome contains thousands of CTCF binding sites [40][41]. Transcription of eRNA may represent a common mechanism allowing neighboring genes to be differentially regulated. A recent paper from Oh et al. showed that regulatory enhancers have the potential to engage more than one promoter in a hosting domain [42]. Different enhancer features, including the level of CTCF, can determine the preference of a specific enhancer in activating different promoters. They referred to this process as “enhancer release and retargeting, ERR” [42]. Importantly, this redirection mechanism of the transcriptional process is at the basis of activation of disease-causing genes [42].5. Noncoding Mutations and Enhancer Hijacking
Germline and somatic single nucleotide variants have been described in enhancers and some of them have been associated with carcinogenesis, inflammatory disorders, cardiomyopathy, and neurodegeneration [32][33][43][44]. Although a portion of them is responsible for alteration of DNA binding by transcription factors [43], this observation enforces the idea that pathogenic mechanisms could involve eRNAs deriving from mutated enhancers [15][33][45]. Among the most common somatic genetic aberrations in cancer, copy number alterations (CNAs) lead to changes in gene dosage of the transcriptional units spanning the altered regions [46]. Chromosomal rearrangements are another common mechanism leading to deregulation of cancer genes, and a possible mechanism is the relocation of regulatory DNA elements including chromatin regions populated with active enhancers, a process dubbed as ‘enhancer hijacking’ [47]. Although MYC overexpression has long been known to occur as result of t(8;14) MYC/IgH gene rearrangement [48], most recently, Zhang et al. showed that duplication of lineage-specific 3′ super-enhancers region to MYC (MYC-LASE) increased MYC expression [49]. Decreased expression of MYC has been observed by CRISPR/KRAB–dCas9 mediated repression or deletion of a constituent enhancer within the MYC-LASE region [49]. Similarly, amplifications of super-enhancers on chromosome segment 13q22.1 are found in different cancer types and activate KLF5 oncogene expression in squamous cell carcinomas [50]. In particular, CRISPR/KRAB–dCas9 mediated multiplexed repression in BICR31 cells of specific enhancers revealed they exert a combinatorial effect on KLF5 activation [50]. In leukemia, intrachromosomal inversion t(3;3)(q21;q26.2) flip GATA2 regulatory element causing EVI1 ectopic activation and GATA2 deregulation, simultaneously [51]. Other chromosomal rearrangements include intrachromosomal deletions of genomic DNA between active enhancers and a proto-oncogene [52][53] and insulator duplication or deletions that are responsible for novel topological and functional chromatin interactions [54]. Most of these studies demonstrate the implication of active SE regions rather than the function of the actual enhancer transcripts. However, there also direct evidence pointing towards eRNAs. Examples are focal somatic CNAs affecting at least one eRNA locus but not protein-coding genes [55] and upregulated eRNAs correlated with CNA [56].6. eRNAs in Cancer
Actively transcribed enhancers regulate most oncogenes and eRNA levels are associated with dysregulated enhancer activation and gene expression in cancer. For example, in sex-hormone-dependent tumors—such as breast and prostate cancers—key regulator eRNAs are induced by hormone receptors and there are many evidences that some eRNAs are prominent factors in sex hormone-dependent cancer development [9][13][57][58][59]. Hormonal regulation of eRNAs might explain the gender association with incidence also in tumors of non-reproductive organs [60]. It is the case of SMAD7e, an estrogen- associated eRNA that contributes to the initiation and progression of bladder cancer [61]. CRISPR-Cas13a technology, applied in bladder cancer cells to downregulate SMAD7e, suppressed proliferation and migration, promoted apoptosis, decreased invasion in bladder cancer, and also impaired the tumor promoting action of estrogen both in vitro and in vivo [61]. Not just sex hormones can modulate enhancer function. Hoffman et al. identified the oncogenic mechanism associated with DDIT4, a gene often dysregulated in a variety of cancer types, showing its modulation by targeting individual regulatory elements within its hormone-responsive super-enhancer [58]. Dexamethasone specifically triggers glucocorticoid receptor binding at different responsive elements in the enhancer, tightly modulating its eRNA expression and therefore regulating DDIT4 transcription [58]. The eRNAs inducing oncogene expression can be considered as potential therapeutic targets, as for instance CCAT1, which directly drives MYC expression in colorectal cancer [62] or other eRNAs specifically transcribed at MYC super-enhancer in hepatocellular carcinoma [63]. In acute myeloid leukemia, dihydroergotamine (DHE)—a drug activator of NR4A nuclear receptors—represses the expression of a selected group of SE-associated leukemic oncogenes including MYC [64]. DHE inhibits MYC SE functional activity by eliminating eRNA transcription and enhancer–promoter looping and suppresses tumor growth both in vitro and in vivo [64]. Vice versa, other eRNAs can be considered as tumor suppressors, such as eRNAs induced by p53 mediating p53-dependent gene transcription [65][66]. Some eRNAs downstream the p53 pathway are not directly bound by p53 but by p53-induced lncRNAs [67]. An example is LED, a lncRNA strongly induced by nutlin-3a, activator of p53. LED can in turn activate p21 eRNA transcription at CDKN1A enhancer and mediate the tumor suppressor function of p53 [67]. Nowadays, a systematic mapping of eRNAs expressed in different types of cancer is available thanks to the effort of scientists who tried to infer the cancer-specific expression of eRNAs from the RNAseq data collected in several cancer series world-wide [32][33][45][55][68]. The expression profile of those eRNAs may help in resolving the intra-tumor heterogeneity and improve the diagnosis and treatment of many cancers. eRNAs are also involved in the development of resistance to anticancer therapies [69]. The cells under treatment can acquire genetic alterations that give them a proliferative advantage, but they can also adapt at an epigenetic level, activating the expression of transcriptional programs that allow cell survival under therapeutic pressure [70][71][72]. Activation of different enhancer networks and eRNA expression are involved in this mechanism [73][74]. eRNAs are clinically relevant because of their cancer-type specific pattern of expression and for such a reason they can represent diagnostic or prognostic markers in cancer therapy [75]. Integrative analysis of multi-omics and pharmacogenomics data across large-scale patient samples and cancer cell lines showed that the majority of genes in the canonical cancer signaling pathways are highly correlated with specific eRNAs in at least one cancer type [76]. The relationship between eRNAs and target genes are confirmed by evidence of chromatin interaction from high-throughput chromosome conformation capture (Hi-C) data. Accordingly, associations between eRNAs and response to anticancer drugs, targeting the linked signaling pathway, emerge crossing data of eRNA expression in 1000 cancer cell lines from Cancer Cell Line Encyclopedia (CCLE), and drug sensitivity available from the Cancer Therapeutics Response Portal (CTRP). A validated example derived from this computational pipeline is NET1e, which is highly expressed in breast cancer and has oncogenic effects in vitro. NET1e in situ overexpression induced drug resistance to BEZ235, a dual PI3K/mTOR inhibitor and obatoclax, pan-BCL2 inhibitor, in breast cancer cell line [76]. Recently, the same research group further improved this approach, estimating the correlation between eRNAs expression with genetic variants, drug response, and immune infiltration in patients to facilitate the functional and clinical investigations of eRNAs in human cancers [45].2. eRNAs Instability and Methodology of Detection
eRNAs are noncoding molecules bidirectionally transcribed from enhancers by RNApol II [15]. The eRNAs are 5′-capped but less stable and shorter than mRNAs because they are retained in the nucleus and exposed to exosome-mediated decay [16]. Indeed, they are mainly unspliced and not polyadenylated. The lack of polyA causes the recruitment of exosome, because the small distance between polyA signal and transcription start site prevents the assembly of polyadenylation machinery at RNApol II C-terminal domain (CTD) [17]. eRNAs are densely located on the chromatin where they help the stabilization of enhancer–promoter (E–P) interactions [17]. These features make them rarely detectable by conventional polyA RNA-seq. Their identification and quantification is feasible by techniques such as ribosomal RNA (rRNA)-depleted total RNA-seq or cap analysis of gene expression (CAGE) [18,19,20]. Other techniques such as the global run-on sequencing (GRO-seq) [21] and its variant precision nuclear run-on sequencing (PRO-seq) [22] investigate RNApol II activity, sequencing the nascent RNA molecules, and thus, they allow the detection of even unstable transcripts. Transient transcriptome sequencing (TT-seq) is often applied to study eRNAs [23,24,25]. It is a sensitive approach to detect short-lived transcripts, which combines a short 4-thiouridine (4sU) RNA labeling pulse with an RNA fragmentation step to enrich for newly synthesized RNA. An adapted CAGE protocol, NET-CAGE, was recently developed to study TSSs of native elongating transcripts (NETs) in a strand-specific manner [26]. It sensitively identifies and quantifies true transcriptional activities of enhancers at high nucleotide resolution and allows the study of RNA synthesis and degradation rates at the TSS level.
Other techniques can help in characterizing eRNA functions, linking the eRNA with its target gene. For instance, an advanced imaging protocol for single molecule fluorescence in situ hybridization (smFISH) was implemented to detect short and lowly expressed transcripts at single cell level [27]. This method uses single biotinylated short (20 nt each) probes, which, post hybridization, allow the binding of multiple copies of fluorophore per probe. The system can be multiplexed to achieve simultaneous detection of both eRNA and the induced nascent gene transcript in the same nucleus to prove their co-localization on the chromatin.
Few technologies are emerging to agnostically infer eRNA function by assessing genome-wide RNA chromatin interactions. RADICL-Seq captures proximal RNA-chromatin interactions in a genome-wide manner [28]. Thanks to the enhancer–promoter looping, this technique can capture the spatial proximity of the nascent RNA with the enhancer region. This improved method overcomes limitations of similar pre-existing techniques such as mapping RNA–genome interactions (MARGI) [29], chromatin-associated RNA-sequencing (CHAR-seq) [30], and global RNA interaction with DNA by deep-sequencing (GRID-seq) [31].
3. eRNAs in Development and Differentiation
The expression of eRNAs is developmental stage and cell-type specific [32,33]. eRNAs transcribed during cellular differentiation are correlated to their target gene expression [2,34] and their production explains the interaction between enhancers and transcription initiation and their proximity to promoters. Enhancer transcription is the most common rapid transcriptional adjustment occurring when cells undergo the first steps toward a state change [19]. Once the target promoter is activated, enhancer activity is no longer required, and so, after a rapid burst of production and activity, eRNAs frequently return to baseline expression levels. In some specific cases, enhancers are rapidly activated and then continuously expressed and these eRNAs might have additional functional roles still unknown [19].
Accordingly, with their pivotal function, eRNAs are involved in determining the developmental stage of cells, often cooperating to regulate the expression of their target genes [2]. General insights into the order of events that establish active enhancers have been recently provided by the application of a genome-wide multi-omics approach to study the C/EBPα-induced trans-differentiation of human precursor leukemia B cells to macrophage-like cells [25]. In particular, TT-seq allowed the identification of transcriptionally active enhancers, then paired with their putative target promoters, and the analysis of changes in transcription activity of enhancers and promoters over time [25]. Most enhancers drive the expression of a common target gene acting in an additive manner, but some enhancers can cooperate synergistically at specific loci to drive target gene transcription [25].
The interacting network of molecules that regulates differentiation is complex and involves different types of lncRNAs. A genome-wide study combined RNA sequencing (RNA-seq) and RNA reverse transcription-associated capture sequencing (RAT-seq) to map interacting targets for lncRNAs [35]. The lcnRNA Peblr20 was identified as a demanding element that helps in maintaining the pluripotent status of induced pluripotent stem cells. Peblr20 knockdown in induced pluripotent stem cells withdrew pluripotency state and, vice versa, overexpression of Peblr20 activated stemness-like genes such as Pou5F1, SOX2, and NANOG in fibroblasts, leading to improved pluripotent reprogramming. Notably, lncRNA Peblr20 recruited TET2 to the Pou5F1 enhancer site, leading to DNA demethylation at the Pou5F1 enhancer and, therefore, to Pou5F1 eRNA expression. Pou5F1 eRNA expression itself seemed to modulate the enhancer promoter looping, activating Pou5F1 expression and induced pluripotent state. Thus, Peblr20 utilizes a novel trans epigenetic eRNA activation mechanism to control the stem cell fate mediated by another ncRNA subgroup, suggesting a widely unexplored interplay between other noncoding regulatory RNAs and eRNAs [35].
4. eRNAs Control Transcription by Several Layers of Sequence Specific Mechanisms
Recently, the abundance of m6A on eRNAs in correspondence of RRACH motif was demonstrated, suggesting an m6A deposition similar to that on mRNAs [36]. Lee at al. showed that the m6A reader YTHDC1 catalyzes the binding of BRD4 to m6A eRNA. In a positive feedback loop, YTHDC1 directly activates enhancers and induces eRNA transcription [37]. However, no evidence of a relationship between m6A and eRNA stability is proven, but on the other hand m6A eRNA bound by YTHDC1 helps chromatin condensation, cross talking to other coactivators and impacting enhancer activity [37]. The act of enhancer transcription generates eRNAs, and the length of these transcripts may influence their m6A levels, consequently facilitating transcriptional activation [37].
Enhancers can be active, primed, or poised, depending on different epigenetic states: active enhancers, which are H3K27ac and H3K4me1 positive and bound by the histone acetyltransferase P300, are strong transcription activators, while, when primed, they have lower H3K27ac and only drive basal transcriptional activation. On the contrary, poised enhancers present H3K4me1 and P300, but they also have the repressive histone mark H3K27me3, associated with Polycomb Repressive Complex 2 (PRC2) silencing [38]. Expression of eRNAs can be activated in poised enhancers by sequence specific mechanism driven by lncRNAs, forming triple-helix structures with DNA [39]. Triplex-based recruitment of chromatin-modifying complexes may represent a common targeting mechanism for enhancer activation. Blank-Giwojna et al. demonstrated that the antisense RNA KHPS1 forms a RNA–DNA triplex at the SPHK1 enhancer, with consequent recruitment of E2F1 and p300 [39]. The enhancer activation induces transcription of an eRNA that is required for SPHK1 expression and cell proliferation. Genomic deletion of the triplex-forming region (TFR) or prevention of KHPS1 binding to DNA impairs cell proliferation and viability [39]. The functional role of the sequence specific lncRNA–DNA binding is proven by the fact that changes in the lncRNA sequence, driving it to distinct genomic location, activating different enhancers and the expression of different genes [39]. The mechanism that leads to promoter activation involves the eRNA-mediated eviction of CTCF, which insulates eSPHK1 from the SPHK1-C promoter [39]. Numerous studies have shown that CTCF regulates enhancer–promoter interactions and the human genome contains thousands of CTCF binding sites [40,41]. Transcription of eRNA may represent a common mechanism allowing neighboring genes to be differentially regulated. A recent paper from Oh et al. showed that regulatory enhancers have the potential to engage more than one promoter in a hosting domain [42]. Different enhancer features, including the level of CTCF, can determine the preference of a specific enhancer in activating different promoters. They referred to this process as “enhancer release and retargeting, ERR” [42]. Importantly, this redirection mechanism of the transcriptional process is at the basis of activation of disease-causing genes [42].
5. Noncoding Mutations and Enhancer Hijacking
Germline and somatic single nucleotide variants have been described in enhancers and some of them have been associated with carcinogenesis, inflammatory disorders, cardiomyopathy, and neurodegeneration [32,33,43,44]. Although a portion of them is responsible for alteration of DNA binding by transcription factors [43], this observation enforces the idea that pathogenic mechanisms could involve eRNAs deriving from mutated enhancers [15,33,45].
Among the most common somatic genetic aberrations in cancer, copy number alterations (CNAs) lead to changes in gene dosage of the transcriptional units spanning the altered regions [46]. Chromosomal rearrangements are another common mechanism leading to deregulation of cancer genes, and a possible mechanism is the relocation of regulatory DNA elements including chromatin regions populated with active enhancers, a process dubbed as ‘enhancer hijacking’ [47].
Although MYC overexpression has long been known to occur as result of t(8;14) MYC/IgH gene rearrangement [48], most recently, Zhang et al. showed that duplication of lineage-specific 3′ super-enhancers region to MYC (MYC-LASE) increased MYC expression [49]. Decreased expression of MYC has been observed by CRISPR/KRAB–dCas9 mediated repression or deletion of a constituent enhancer within the MYC-LASE region [49]. Similarly, amplifications of super-enhancers on chromosome segment 13q22.1 are found in different cancer types and activate KLF5 oncogene expression in squamous cell carcinomas [50]. In particular, CRISPR/KRAB–dCas9 mediated multiplexed repression in BICR31 cells of specific enhancers revealed they exert a combinatorial effect on KLF5 activation [50]. In leukemia, intrachromosomal inversion t(3;3)(q21;q26.2) flip GATA2 regulatory element causing EVI1 ectopic activation and GATA2 deregulation, simultaneously [51]. Other chromosomal rearrangements include intrachromosomal deletions of genomic DNA between active enhancers and a proto-oncogene [52,53] and insulator duplication or deletions that are responsible for novel topological and functional chromatin interactions [54].
Most of these studies demonstrate the implication of active SE regions rather than the function of the actual enhancer transcripts. However, there also direct evidence pointing towards eRNAs. Examples are focal somatic CNAs affecting at least one eRNA locus but not protein-coding genes [55] and upregulated eRNAs correlated with CNA [56].
6. eRNAs in Cancer
Actively transcribed enhancers regulate most oncogenes and eRNA levels are associated with dysregulated enhancer activation and gene expression in cancer. For example, in sex-hormone-dependent tumors—such as breast and prostate cancers—key regulator eRNAs are induced by hormone receptors and there are many evidences that some eRNAs are prominent factors in sex hormone-dependent cancer development [9,13,57,58,59]. Hormonal regulation of eRNAs might explain the gender association with incidence also in tumors of non-reproductive organs [60]. It is the case of SMAD7e, an estrogen- associated eRNA that contributes to the initiation and progression of bladder cancer [61]. CRISPR-Cas13a technology, applied in bladder cancer cells to downregulate SMAD7e, suppressed proliferation and migration, promoted apoptosis, decreased invasion in bladder cancer, and also impaired the tumor promoting action of estrogen both in vitro and in vivo [61]. Not just sex hormones can modulate enhancer function. Hoffman et al. identified the oncogenic mechanism associated with DDIT4, a gene often dysregulated in a variety of cancer types, showing its modulation by targeting individual regulatory elements within its hormone-responsive super-enhancer [58]. Dexamethasone specifically triggers glucocorticoid receptor binding at different responsive elements in the enhancer, tightly modulating its eRNA expression and therefore regulating DDIT4 transcription [58].
The eRNAs inducing oncogene expression can be considered as potential therapeutic targets, as for instance CCAT1, which directly drives MYC expression in colorectal cancer [62] or other eRNAs specifically transcribed at MYC super-enhancer in hepatocellular carcinoma [63]. In acute myeloid leukemia, dihydroergotamine (DHE)—a drug activator of NR4A nuclear receptors—represses the expression of a selected group of SE-associated leukemic oncogenes including MYC [64]. DHE inhibits MYC SE functional activity by eliminating eRNA transcription and enhancer–promoter looping and suppresses tumor growth both in vitro and in vivo [64]. Vice versa, other eRNAs can be considered as tumor suppressors, such as eRNAs induced by p53 mediating p53-dependent gene transcription [65,66]. Some eRNAs downstream the p53 pathway are not directly bound by p53 but by p53-induced lncRNAs [67]. An example is LED, a lncRNA strongly induced by nutlin-3a, activator of p53. LED can in turn activate p21 eRNA transcription at CDKN1A enhancer and mediate the tumor suppressor function of p53 [67].
Nowadays, a systematic mapping of eRNAs expressed in different types of cancer is available thanks to the effort of scientists who tried to infer the cancer-specific expression of eRNAs from the RNAseq data collected in several cancer series world-wide [32,33,45,55,68]. The expression profile of those eRNAs may help in resolving the intra-tumor heterogeneity and improve the diagnosis and treatment of many cancers.
eRNAs are also involved in the development of resistance to anticancer therapies [69]. The cells under treatment can acquire genetic alterations that give them a proliferative advantage, but they can also adapt at an epigenetic level, activating the expression of transcriptional programs that allow cell survival under therapeutic pressure [70,71,72]. Activation of different enhancer networks and eRNA expression are involved in this mechanism [73,74]. eRNAs are clinically relevant because of their cancer-type specific pattern of expression and for such a reason they can represent diagnostic or prognostic markers in cancer therapy [75]. Integrative analysis of multi-omics and pharmacogenomics data across large-scale patient samples and cancer cell lines showed that the majority of genes in the canonical cancer signaling pathways are highly correlated with specific eRNAs in at least one cancer type [76]. The relationship between eRNAs and target genes are confirmed by evidence of chromatin interaction from high-throughput chromosome conformation capture (Hi-C) data. Accordingly, associations between eRNAs and response to anticancer drugs, targeting the linked signaling pathway, emerge crossing data of eRNA expression in 1000 cancer cell lines from Cancer Cell Line Encyclopedia (CCLE), and drug sensitivity available from the Cancer Therapeutics Response Portal (CTRP). A validated example derived from this computational pipeline is NET1e, which is highly expressed in breast cancer and has oncogenic effects in vitro. NET1e in situ overexpression induced drug resistance to BEZ235, a dual PI3K/mTOR inhibitor and obatoclax, pan-BCL2 inhibitor, in breast cancer cell line [76]. Recently, the same research group further improved this approach, estimating the correlation between eRNAs expression with genetic variants, drug response, and immune infiltration in patients to facilitate the functional and clinical investigations of eRNAs in human cancers [45].