Colloidal metal nanoparticles in an electrolyte environment are not only electrically charged but also electrochemically active objects. They have the typical character of metal electrodes with ongoing charge transfer processes on the metal/liquid interface. This picture is valid for the equilibrium state and also during the formation, growth, aggregation or dissolution of nanoparticles. This behavior can be understood in analogy to macroscopic mixed-electrode systems with a free-floating potential, which is determined by the competition between anodic and cathodic partial processes. In contrast to macroscopic electrodes, the small size of nanoparticles is responsible for significant effects of low numbers of elementary charges and for self-polarization effects as they are known from molecular systems, for example. The electrical properties of nanoparticles can be estimated by basic electrochemical equations. Reconsidering these fundamentals, the assembly behavior, the formation of nonspherical assemblies of nanoparticles and the growth and the corrosion behavior of metal nanoparticles, as well as the formation of core/shell particles, branched structures and particle networks, can be understood. The consequences of electrochemical behavior, charging and self-polarization for particle growth, shape formation and particle/particle interaction are discussed.
- nanoparticles
- colloidal solutions
- electrical charging
- self-polarization
- mixed-electrode
- particle growth
- particle interaction
1. Introduction
2. Formation of Colloidal Metal Nanoparticles by Reduction of Solution Precursors
3. Forming Colloidal Metal Nanoparticles as Mixed-Electrode Objects
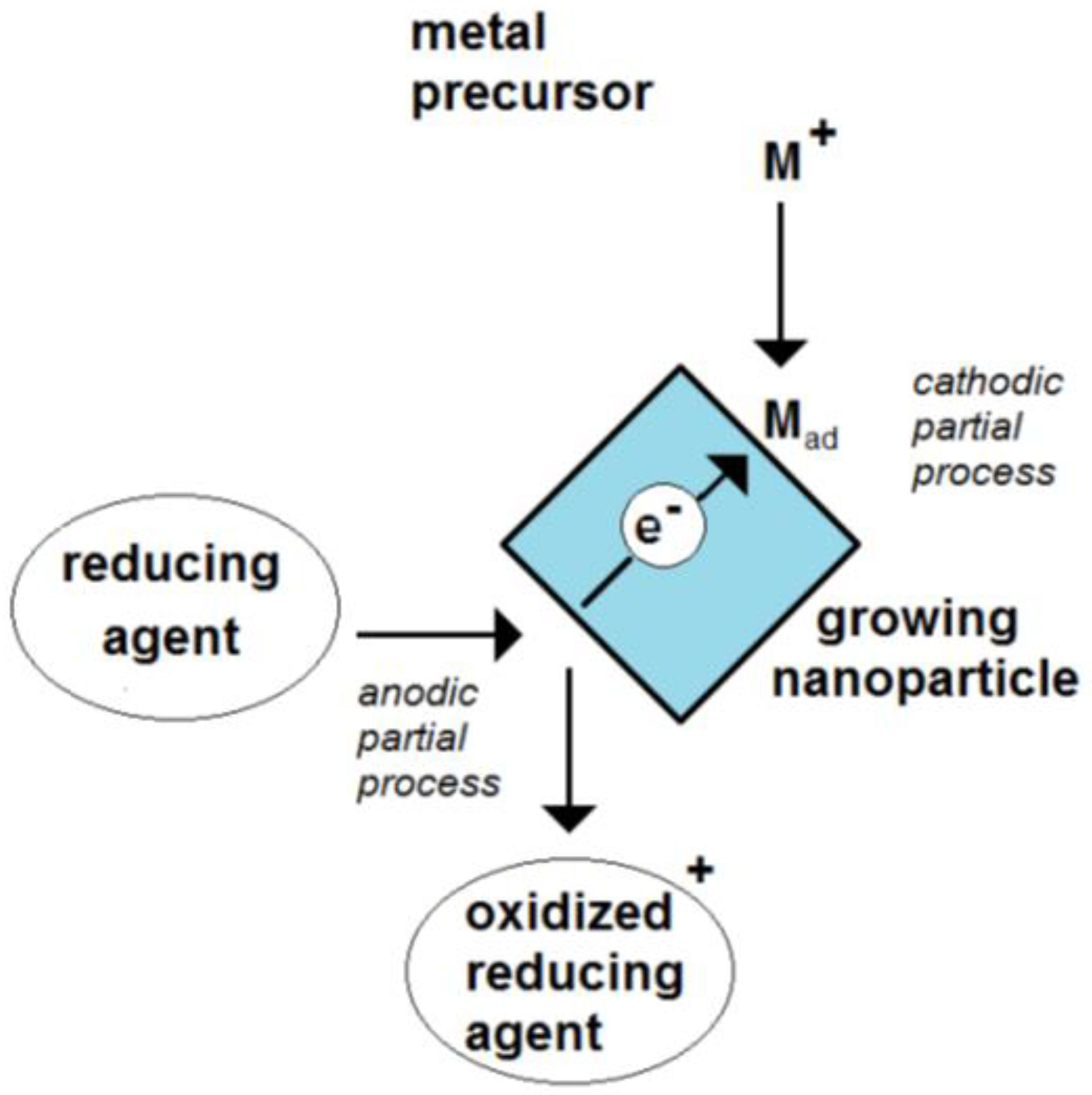 Figure 1. Scheme of the formation of an electrochemical mixed potential of a growing nanoparticle by the coupling of a cathodic and an anodic partial process.
Figure 1. Scheme of the formation of an electrochemical mixed potential of a growing nanoparticle by the coupling of a cathodic and an anodic partial process.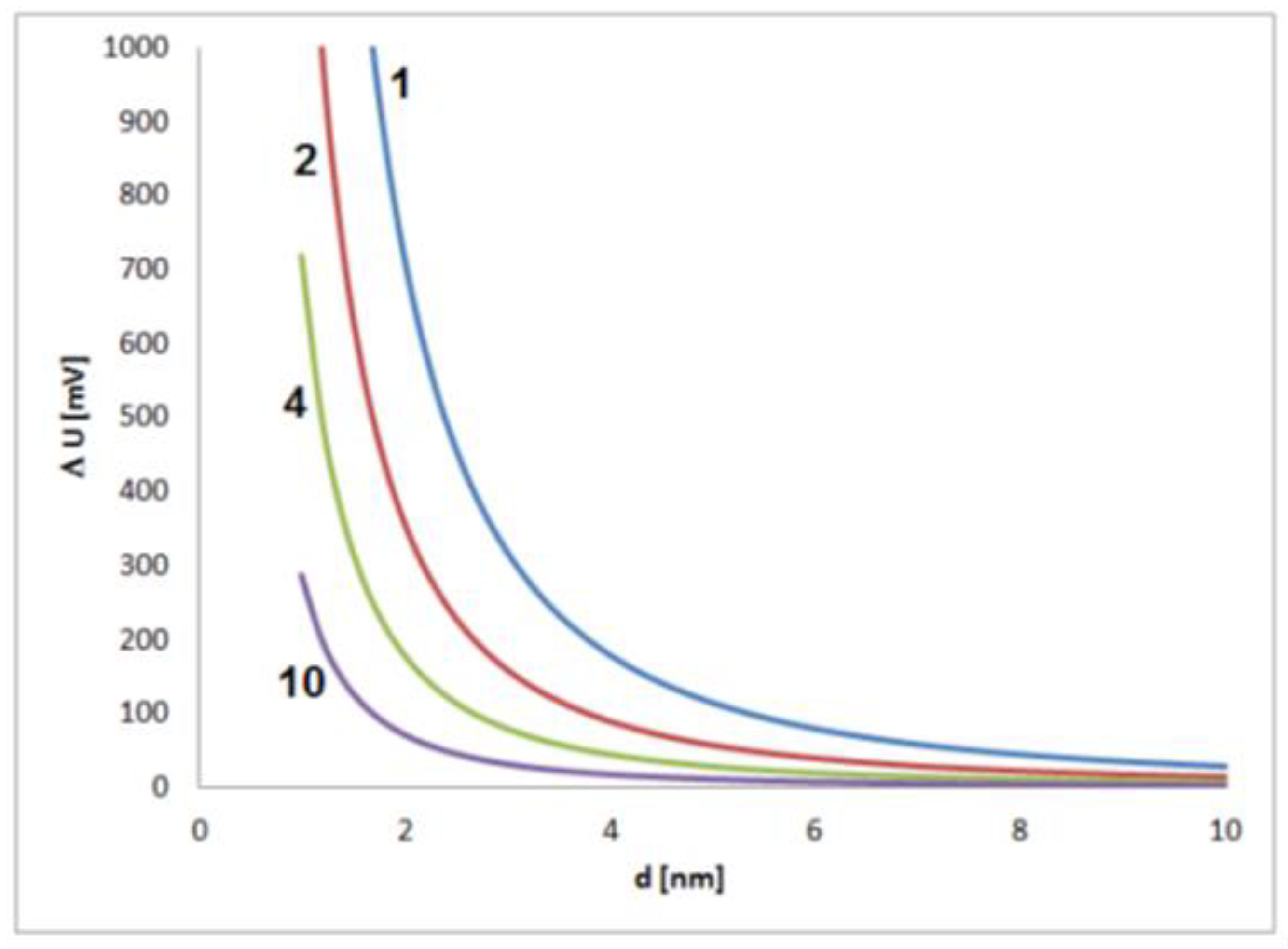 Figure 2. Estimated contribution of a single elementary charge on the potential of a single metal nanoparticle in dependence on the particle size for values of dielectric constant (electrical permittivity) of 1, 2, 4 and 10 and an assumed thickness of the electrochemical double layer of 0.5 nm.
Figure 2. Estimated contribution of a single elementary charge on the potential of a single metal nanoparticle in dependence on the particle size for values of dielectric constant (electrical permittivity) of 1, 2, 4 and 10 and an assumed thickness of the electrochemical double layer of 0.5 nm.4. Self-Polarization Effects of Nonspherical Metal Nanoparticles
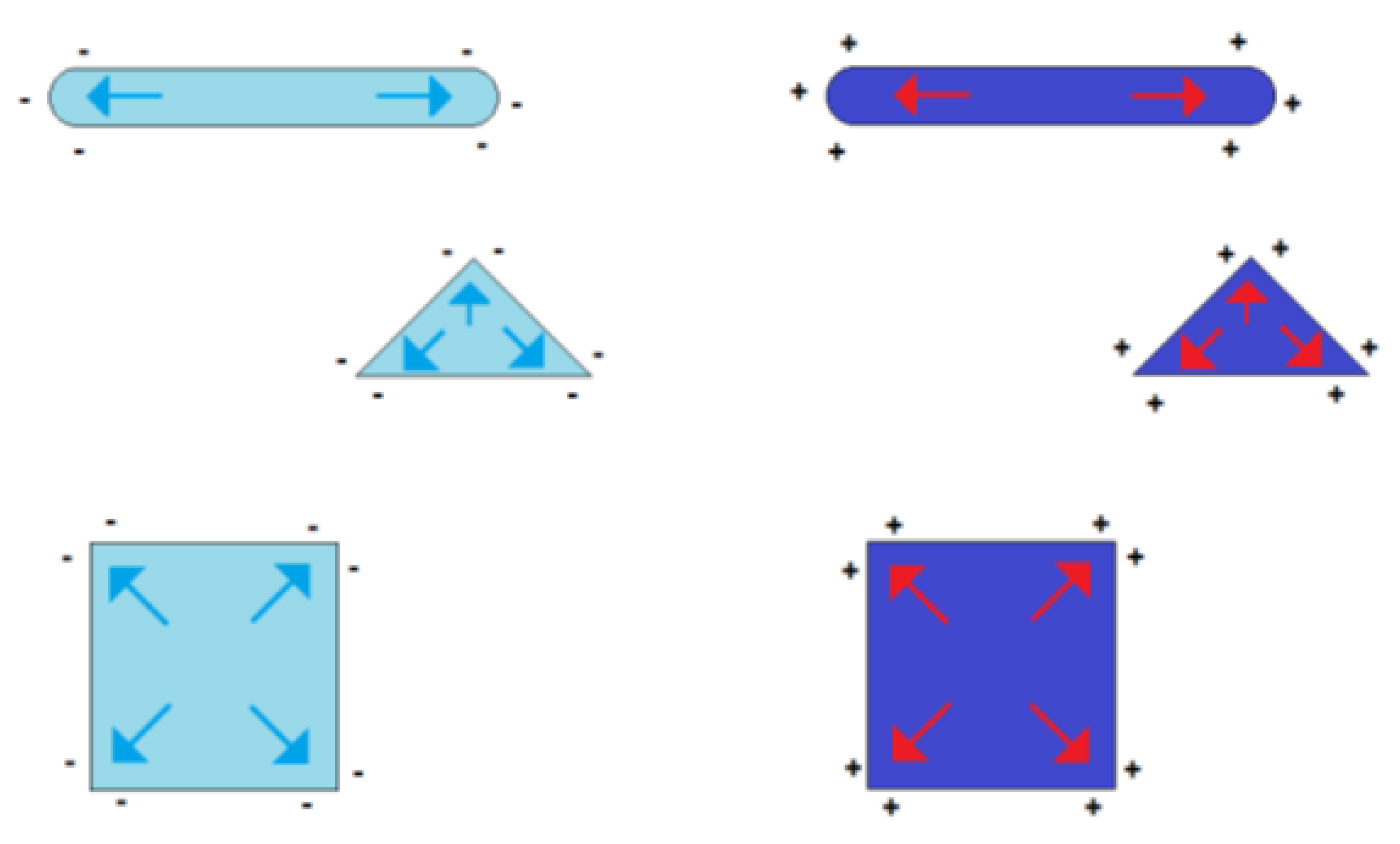 Figure 3. Formation of charge centers of gravity in the case of electrically charged nonspherical metal nanoparticles (blue symbolizing negative excess charge, red symbolizing positive excess charge).
Figure 3. Formation of charge centers of gravity in the case of electrically charged nonspherical metal nanoparticles (blue symbolizing negative excess charge, red symbolizing positive excess charge).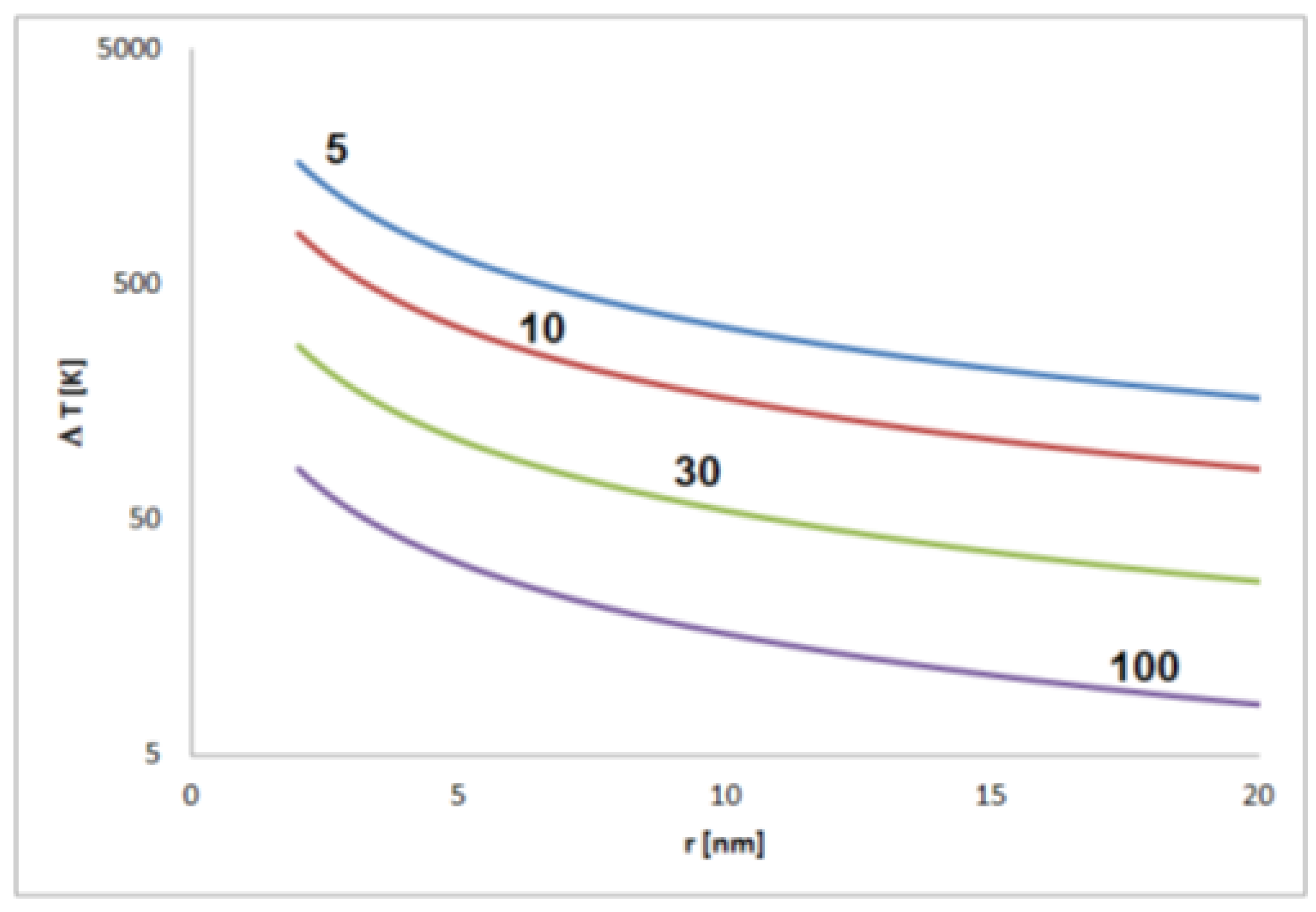 Figure 4. Estimated temperature differences T corresponding to the thermal energy equal to the electrostatic energy of a pair of elementary charges in distance r (for permittivity of 5, 10, 30 and 100).
Figure 4. Estimated temperature differences T corresponding to the thermal energy equal to the electrostatic energy of a pair of elementary charges in distance r (for permittivity of 5, 10, 30 and 100).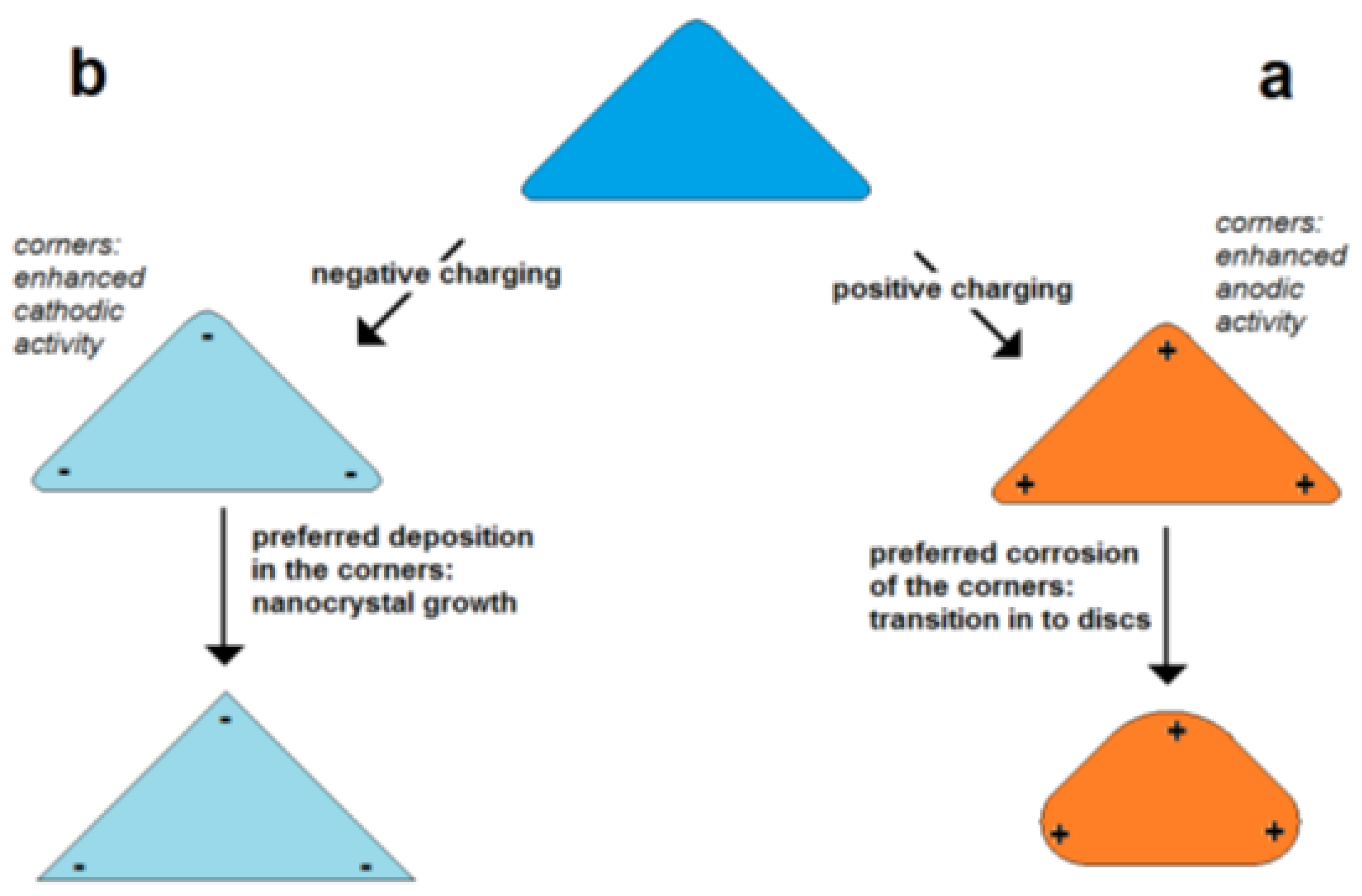 Figure 5. Self-polarization effect: effect of excess charge on the change of particle shape in dependence on charge sign shown for the example of a flat triangular metal nanoprism (schematically); (a) enhanced positive charge in the corners of triangular particle, (b) enhanced negative charge in the corners of particles.
Figure 5. Self-polarization effect: effect of excess charge on the change of particle shape in dependence on charge sign shown for the example of a flat triangular metal nanoprism (schematically); (a) enhanced positive charge in the corners of triangular particle, (b) enhanced negative charge in the corners of particles.5. Factors in Shape Control of Growing Metal Nanoparticles
6. Electrically Controlled Assembly of Metal Nanoparticles
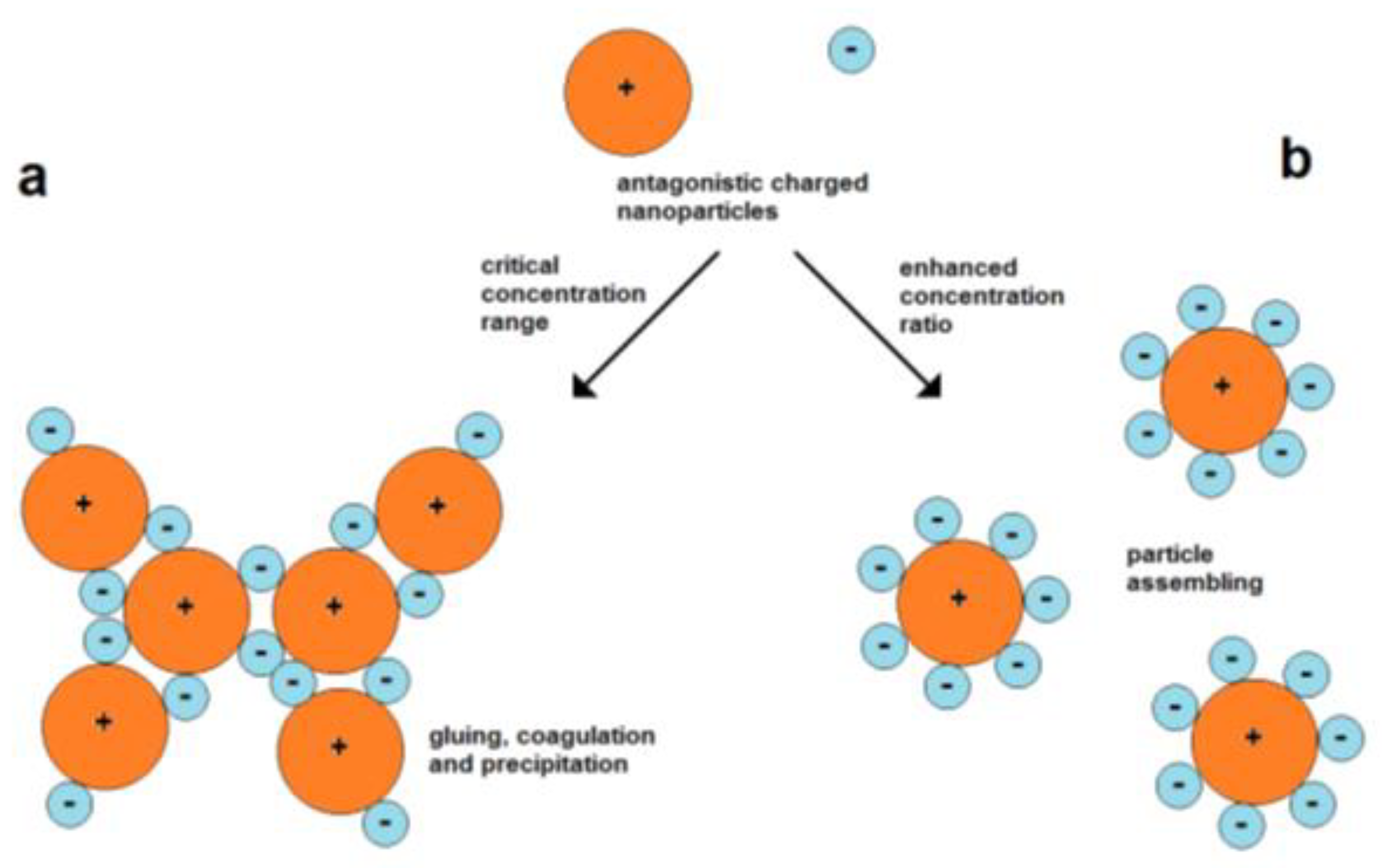 Figure 6. Coagulation and assembly of oppositely charged nanoparticles; (a) high probability of coagulation at lower concentration ratios, (b) forming of dispersed assemblies at higher concentration ratios between small negative and larger positive particles.
Figure 6. Coagulation and assembly of oppositely charged nanoparticles; (a) high probability of coagulation at lower concentration ratios, (b) forming of dispersed assemblies at higher concentration ratios between small negative and larger positive particles.-
Raising the general ion concentrations, which means the ionic strength (“salt-out effect”);
-
Targeted addition of antagonistic ions, which means metal cations with high surface affinity in the case of negatively charged particles;
-
Targeted addition of surface-affine anions or electron-donor ligands in the case of positively charged particles;
-
Lowering the electrochemical potential of particles in the case of positively charged particles by enhancing the concentration of reducing agents;
-
Raising the electrochemical potential in the case of negatively charged particles by the addition of oxidizing agents.
7. Secondary Metal Deposition
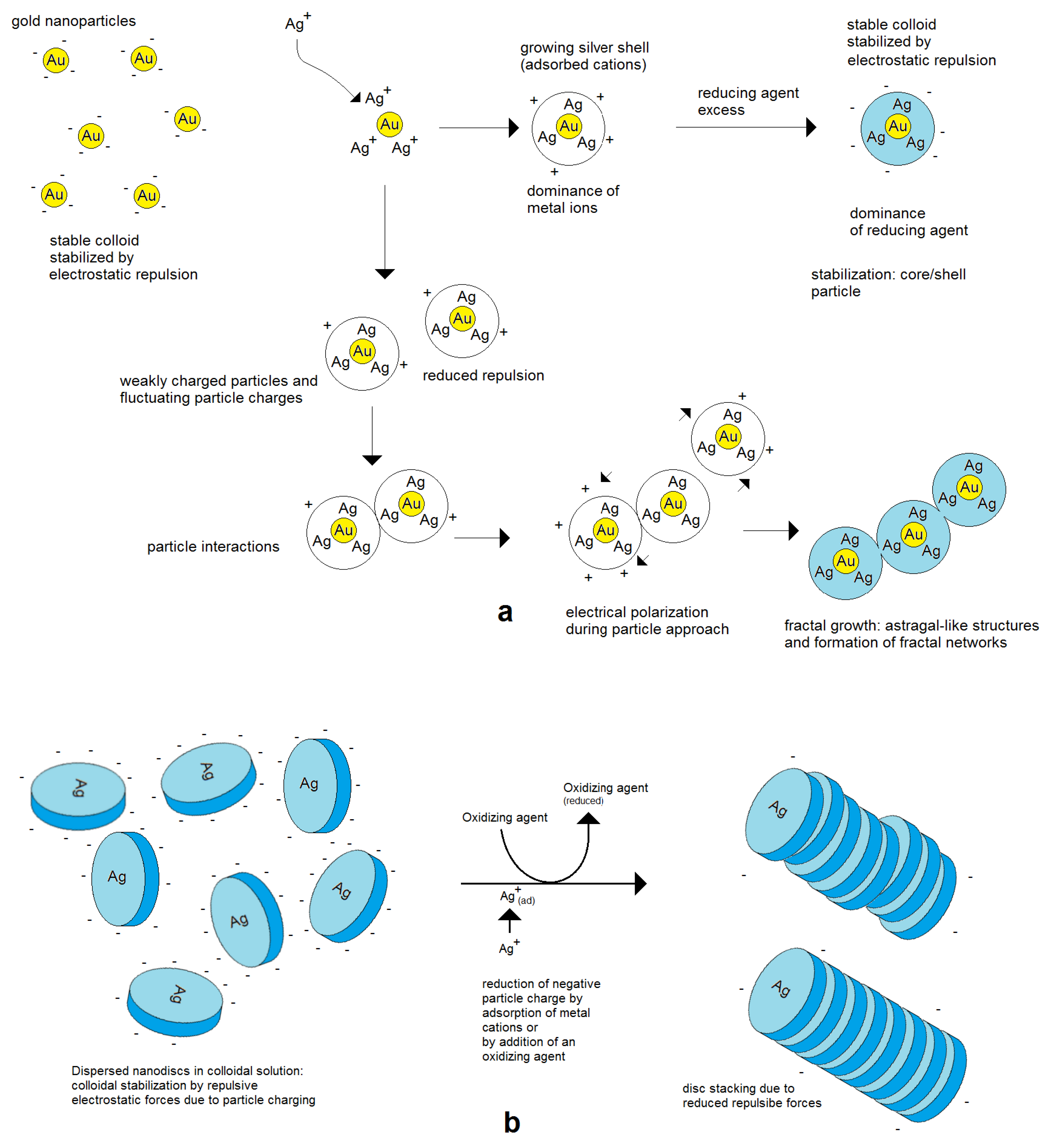 Figure 7. Assembly behavior of charged metal nanoparticles: (a) spherical nanoparticles; (b) nonspherical metal nanoparticles, for example nanodiscs.
Figure 7. Assembly behavior of charged metal nanoparticles: (a) spherical nanoparticles; (b) nonspherical metal nanoparticles, for example nanodiscs.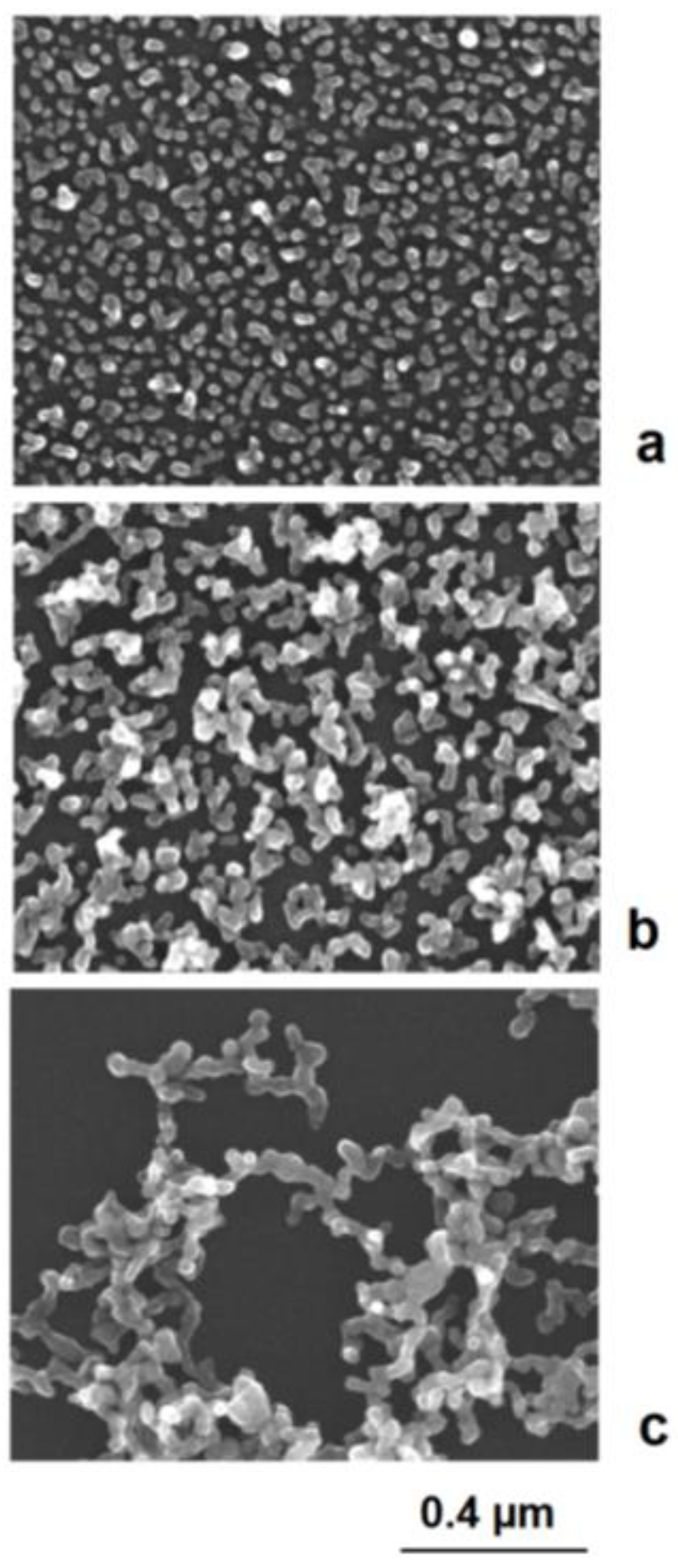 Figure 8. In situ formation of aggregate particles during the chemical deposition of silver nanoparticles on gold seeds: (a) small aggregate particles; (b) enhanced aggregate size; (c) network formation.
Figure 8. In situ formation of aggregate particles during the chemical deposition of silver nanoparticles on gold seeds: (a) small aggregate particles; (b) enhanced aggregate size; (c) network formation.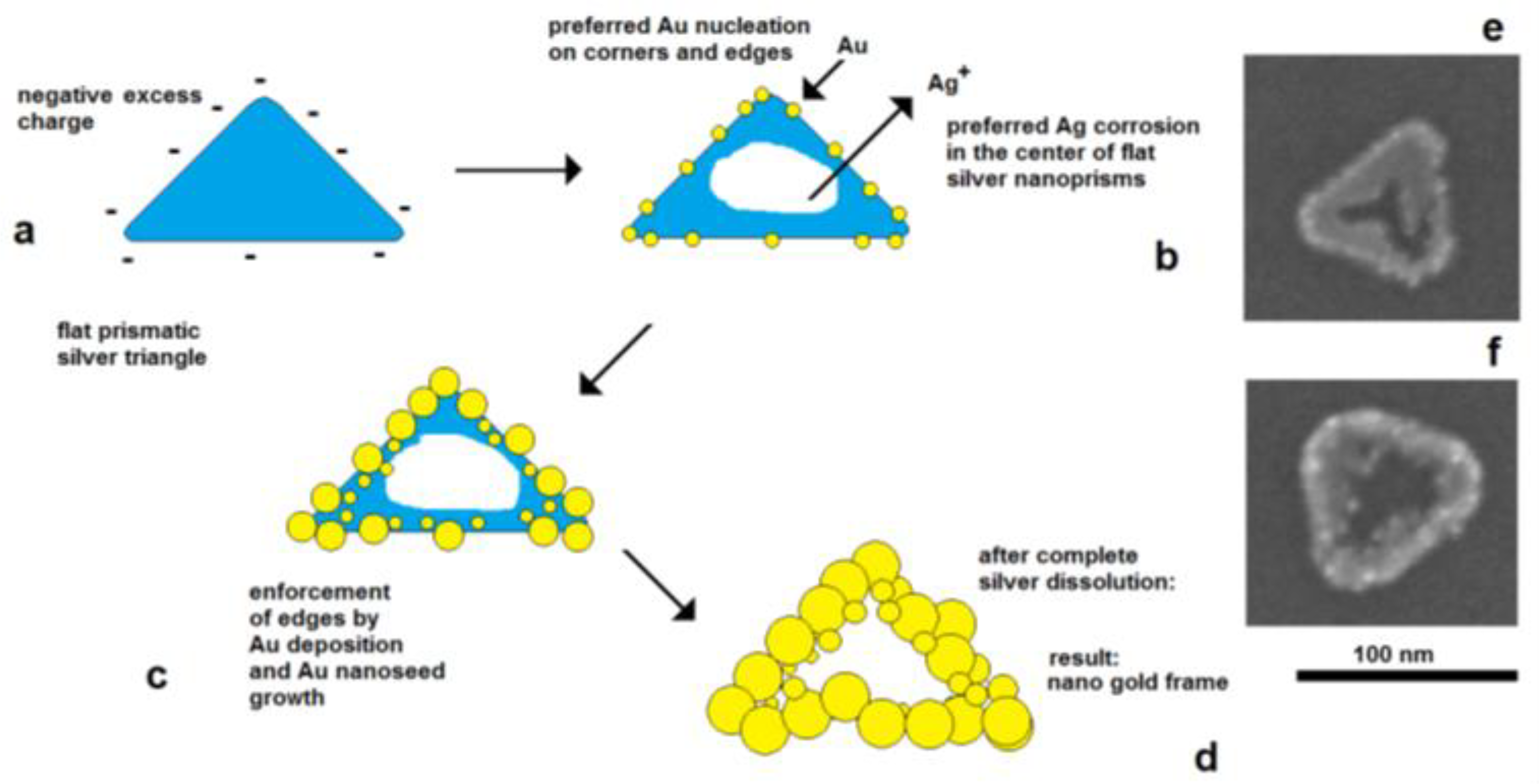 Figure 9. Electrically controlled formation of metal frame particles by the substitution of a less-noble metal by a noble metal. Deposition of gold on flat silver nanotriangles: (a) negatively charged silver nanotriangles in the colloidal state; (b) starting gold deposition on the particle edges (lowest potential) and starting silver corrosion in the center of the flat prism (highest potential); (c) enforcement of gold in the edge regions of particles; (d) formation of a gold frame by the complete dissolution of silver (schematically); (e) silver triangle with a hole formation in the particle center; (f) advanced silver dissolution and edge-directed gold deposition (SEM images).
Figure 9. Electrically controlled formation of metal frame particles by the substitution of a less-noble metal by a noble metal. Deposition of gold on flat silver nanotriangles: (a) negatively charged silver nanotriangles in the colloidal state; (b) starting gold deposition on the particle edges (lowest potential) and starting silver corrosion in the center of the flat prism (highest potential); (c) enforcement of gold in the edge regions of particles; (d) formation of a gold frame by the complete dissolution of silver (schematically); (e) silver triangle with a hole formation in the particle center; (f) advanced silver dissolution and edge-directed gold deposition (SEM images).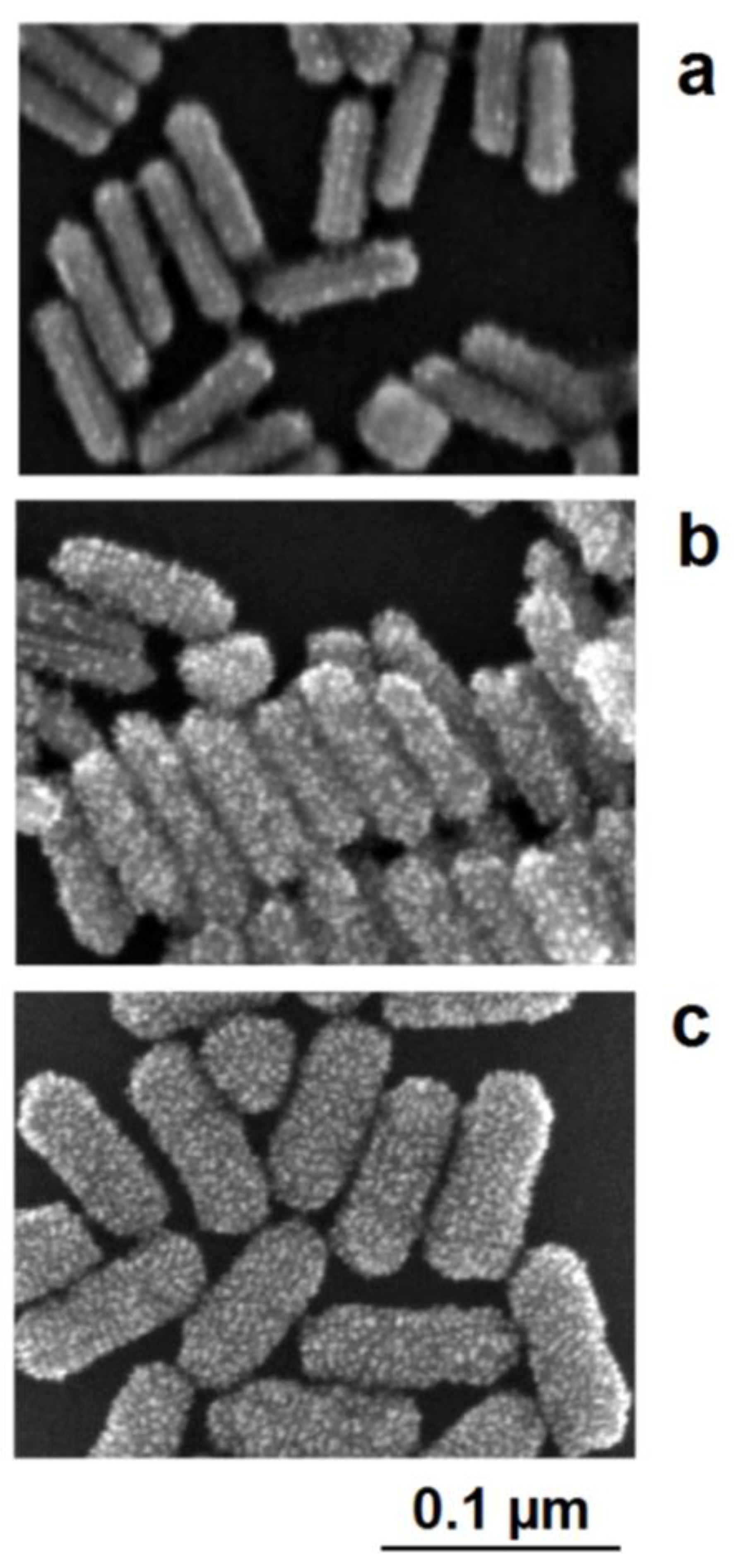 Figure 10. Interplay between the lattice-controlled and accidental nucleation of platinum on gold nanorods: (a) lowest platinum precursor concentration: decoration of borderlines between crystallographic planes; (b) mediate precursor concentration: additional nucleation on the crystallographic planes between the excellent borderlines; (c) high platinum precursor concentration: densified platinum deposition as a shell around the gold core.
Figure 10. Interplay between the lattice-controlled and accidental nucleation of platinum on gold nanorods: (a) lowest platinum precursor concentration: decoration of borderlines between crystallographic planes; (b) mediate precursor concentration: additional nucleation on the crystallographic planes between the excellent borderlines; (c) high platinum precursor concentration: densified platinum deposition as a shell around the gold core.8. Conclusions
References
- Homberger, M.; Simon, U. On the application potential of gold nanoparticles in nanoelectronics and biomedicine. Philos. Trans. R. Soc. A-Math. Phys. Eng. Sci. 2010, 368, 1405–1453.Homberger, M.; Simon, U. On the application potential of gold nanoparticles in nanoelectronics and biomedicine. Philos. Trans. R. Soc. A-Math. Phys. Eng. Sci. 2010, 368, 1405–1453.
- Link, S.; El-Sayed, M.A. Spectral properties and relaxation dynamics of surface plasmon electronic oscillations in gold and silver nanodots and nanorods. J. Phys. Chem. B 1999, 103, 8410–8426.Link, S.; El-Sayed, M.A. Spectral properties and relaxation dynamics of surface plasmon electronic oscillations in gold and silver nanodots and nanorods. J. Phys. Chem. B 1999, 103, 8410–8426.
- Senthil Kumar, P.; Pastoriza-Santos, I.; Rodriguez-Gonzalez, B.; Garcia de Abajo, F.J.; Liz-Marzan, L.M. High-yield synthesis and optical response of gold nanostars. Nanotechnology 2008, 19, 015606.Senthil Kumar, P.; Pastoriza-Santos, I.; Rodriguez-Gonzalez, B.; Garcia de Abajo, F.J.; Liz-Marzan, L.M. High-yield synthesis and optical response of gold nanostars. Nanotechnology 2008, 19, 015606.
- Amendola, V.; Pilot, R.; Frasconi, M.; Maragò, O.M.; Iatì, M.A. Surface plasmon resonance in gold nanoparticles: A review. J. Phys. 2017, 29, 203002.Amendola, V.; Pilot, R.; Frasconi, M.; Maragò, O.M.; Iatì, M.A. Surface plasmon resonance in gold nanoparticles: A review. J. Phys. 2017, 29, 203002.
- Reichert, J.; Csaki, A.; Kohler, J.M.; Fritzsche, W. Chip-based optical detection of DNA hybridization by means of nanobead labeling. Anal. Chem. 2000, 72, 6025–6029.Reichert, J.; Csaki, A.; Kohler, J.M.; Fritzsche, W. Chip-based optical detection of DNA hybridization by means of nanobead labeling. Anal. Chem. 2000, 72, 6025–6029.
- Csaki, A.; Kaplanek, P.; Moller, R.; Fritzsche, W. The optical detection of individual DNA-conjugated gold nanoparticle labels after metal enhancement. Nanotechnology 2003, 14, 1262–1268.Csaki, A.; Kaplanek, P.; Moller, R.; Fritzsche, W. The optical detection of individual DNA-conjugated gold nanoparticle labels after metal enhancement. Nanotechnology 2003, 14, 1262–1268.
- Penn, S.G.; He, L.; Natan, M.J. Nanoparticles for bioanalysis. Curr. Opin. Chem. Biol. 2003, 7, 609–615.Penn, S.G.; He, L.; Natan, M.J. Nanoparticles for bioanalysis. Curr. Opin. Chem. Biol. 2003, 7, 609–615.
- Khoury, C.G.; Vo-Dinh, T. Gold Nanostars For Surface-Enhanced Raman Scattering: Synthesis, Characterization and Optimization. J. Phys. Chem. C 2008, 112, 18849–18859.Khoury, C.G.; Vo-Dinh, T. Gold Nanostars For Surface-Enhanced Raman Scattering: Synthesis, Characterization and Optimization. J. Phys. Chem. C 2008, 112, 18849–18859.
- Köhler, J.M.; März, A.; Popp, J.; Knauer, A.; Kraus, I.; Faerber, J.; Serra, C. Polyacrylamide/silver composite particles produced via microfluidic photopolymerization for single particle-based SERS microsensorics. Anal. Chem. 2013, 85, 313–318.Köhler, J.M.; März, A.; Popp, J.; Knauer, A.; Kraus, I.; Faerber, J.; Serra, C. Polyacrylamide/silver composite particles produced via microfluidic photopolymerization for single particle-based SERS microsensorics. Anal. Chem. 2013, 85, 313–318.
- Gao, Z.; Ye, H.; Tang, D.; Tao, J.; Habibi, S.; Minerick, A.; Tang, D.; Xia, X. Platinum-decorated gold nanoparticles with dual functionalities for ultrsensitive colorimetric in vitro diagnostics. Nano Lett. 2017, 17, 5572–5579.Gao, Z.; Ye, H.; Tang, D.; Tao, J.; Habibi, S.; Minerick, A.; Tang, D.; Xia, X. Platinum-decorated gold nanoparticles with dual functionalities for ultrsensitive colorimetric in vitro diagnostics. Nano Lett. 2017, 17, 5572–5579.
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079.Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079.
- Kawahara, K.; Inoue-Kahino, N.; Namie, K.; Kato, Y.; Tomo, T.; Shibata, Y.; Kashino, Y.; Noguchi, T. A gold nanoparticle conjugate with photosystem I and photosystem II for development of a biohybrid water-splitting photocatalyst. Biomed. Spectrosc. Imaging 2020, 9, 73–81.Kawahara, K.; Inoue-Kahino, N.; Namie, K.; Kato, Y.; Tomo, T.; Shibata, Y.; Kashino, Y.; Noguchi, T. A gold nanoparticle conjugate with photosystem I and photosystem II for development of a biohybrid water-splitting photocatalyst. Biomed. Spectrosc. Imaging 2020, 9, 73–81.
- Haes, A.J.; Zou, S.L.; Schatz, G.C.; van Duyne, R.P. Nanoscale optical biosensor: Short range distance dependence of the localized surface plasmon resonance of noble metal nanoparticles. J. Phys. Chem. B 2004, 108, 6961–6968.Haes, A.J.; Zou, S.L.; Schatz, G.C.; van Duyne, R.P. Nanoscale optical biosensor: Short range distance dependence of the localized surface plasmon resonance of noble metal nanoparticles. J. Phys. Chem. B 2004, 108, 6961–6968.
- Kong, F.-Y.; Zhang, J.-W.; Li, R.-F. Unique role of gold nanoparticles in drug delivery, targeting and imaging applications. Molecules 2017, 22, 1445.Kong, F.-Y.; Zhang, J.-W.; Li, R.-F. Unique role of gold nanoparticles in drug delivery, targeting and imaging applications. Molecules 2017, 22, 1445.
- Alim, S.; Vejavan, J.; Yusoff, M.; Kafi, A.K.M. Recent use of carbon nanotubes & gold nanoparticles in electrochemistry with applications in biosensing: A review. Biosens. Bioelectron. 2018, 121, 125–136.Alim, S.; Vejavan, J.; Yusoff, M.; Kafi, A.K.M. Recent use of carbon nanotubes & gold nanoparticles in electrochemistry with applications in biosensing: A review. Biosens. Bioelectron. 2018, 121, 125–136.
- Liu, N.; Liedl, T. DNA-assembled advanced plasmonic architectures. Chem. Rev. 2018, 118, 3032–3053.Liu, N.; Liedl, T. DNA-assembled advanced plasmonic architectures. Chem. Rev. 2018, 118, 3032–3053.
- Singh, R.; Belgamwar, R.; Dhiman, M.; Polshettiwar, V. Dendritic fibrous nano-silica-supported gold nanoparticles as an artificial enzyme. J. Mater. Chem. B 2018, 6, 1600–1604.Singh, R.; Belgamwar, R.; Dhiman, M.; Polshettiwar, V. Dendritic fibrous nano-silica-supported gold nanoparticles as an artificial enzyme. J. Mater. Chem. B 2018, 6, 1600–1604.
- Li, G.; Zhao, S.; Zhang, Y.; Tang, Z. Metal–Organic Frameworks Encapsulating Active Nanoparticles as Emerging Compo-sites for Catalysis: Recent Progress and Perspectives. Adv. Mater. 2018.Li, G.; Zhao, S.; Zhang, Y.; Tang, Z. Metal–Organic Frameworks Encapsulating Active Nanoparticles as Emerging Compo-sites for Catalysis: Recent Progress and Perspectives. Adv. Mater. 2018.
- Capek, I. Noble metal nanoparticles: Preparation, composite nanostructures, biodecoration and collective properties. Nanostructure Sci. Technol. 2017, 30, 211–316.Capek, I. Noble metal nanoparticles: Preparation, composite nanostructures, biodecoration and collective properties. Nanostructure Sci. Technol. 2017, 30, 211–316.
- Lee, S.H.; Jun, B.-H. Silver nanoparticles: Synthesis and application for nanomedicine. Int. J. Mol. Sci. 2019, 20, 865.Lee, S.H.; Jun, B.-H. Silver nanoparticles: Synthesis and application for nanomedicine. Int. J. Mol. Sci. 2019, 20, 865.
- Koehler, J.M.; Visaveliya, N.; Knauer, A. Controlling formation and assembling of nanoparticles by control of electrical charging, polarization, and electrochemical potential. Nanotechnol. Rev. 2014, 3, 553–568.Koehler, J.M.; Visaveliya, N.; Knauer, A. Controlling formation and assembling of nanoparticles by control of electrical charging, polarization, and electrochemical potential. Nanotechnol. Rev. 2014, 3, 553–568.
- Whitehead, C.B.; Saim, O.; Finke, R.G. LaMer’s 1950 model of particle formation: A review and critical analysis of its classical nucleation and fluctuation theory basis, of competing models and mechanisms for phase-changes and particle formation, and then of its application to silver halide, semiconductor, metal, and metal-oxide nanoparticles. Mater. Adv. 2021, 2, 186–235.
- Polte, J.; Ahner, T.T.; Delissen, F.; Sokolov, S.; Emmerling, F.; Thuenemann, A.F.; Kraehnert, R. Mechanism of Gold Nanoparticle Formation in the Classical Citrate Synthesis Method Derived from Coupled In Situ XANES and SAXS Evaluation. J. Am. Chem. Soc. 2010, 132, 1296–1301.
- Polte, J.; Erler, R.; Thuenemann, A.F.; Sokolov, S.; Ahner, T.T.; Rademann, K.; Emmerling, F.; Kraehnert, R. Nucleation and growth of gold nanoparticles studied via in situ Small Angle X-ray Scattering at millisecond time resolution. ACS Nano 2010, 4, 1076–1082.
- Sohn, K.; Kim, F.; Pradel, K.C.; Peng, Y.; Zhou, F.; Huang, J. Construction of evolutionary tree for morphological engineering of nanoparticles. ACS Nano 2009, 3, 2191–2198.
- Xia, Y.; Xia, X.; Peng, H.-C. Shape-controlled synthesis of colloidal metal nanocrystals: Thermodynamic versus kinetic products. JACS 2015, 137, 7947–7966.
- Chen, Z.; Balankura, T.; Fichthorn, K.A.; Rioux, R.M. Revisiting the polyol synthesis of silver nanostructures. Role of chloride in nanocube formation. ACS Nano 2019, 13, 1849–1860.
- Khanal, B.P.; Zubarev, E.R. Chemical transformation of nanorods to nanowires: Reversible growth and dissolution of anisotropic gold nanostructures. ACS Nano 2019, 13, 2370–2378.
- Khlebtsov, B.N.; Khanadeev, V.A.; Ye, J.; Sukhourukov, G.B.; Khlebtsov, N.G. Overgrowth of gold nanorods by using a binary surfactant mixture. Langmuir 2014, 30, 1696–1703.
- Sau, T.K.; Murphy, C.J. Seeded high yield synthesis of short Au nanorods in aqueous solution. Langmuir 2004, 20, 6414–6420.
- Köhler, J.M.; Romanus, H.; Hübner, U.; Wagner, J. Formation of star-like and core-shell AuAg nanoparticles during two- and three-step preparation in batch and in microfluidic systems. J. Nanomater. 2007, 98134.
- Novo, C.; Mulvaney, P. Charge-induced Rayleigh instabilities in small gold nanorods. Nano Lett. 2007, 7, 520–524.
- Visaveliya, N.; Knauer, A.; Köhler, J.M. Application of polyionic macromolecules in micro flow syntheses of nanoparticles. Macromol. Chem. Phys. 2017, 218, 1600371.
- Koehler, J.M.; Kluitmann, J.; Knauer, A. Metal Nano Networks by Potential-Controlled In Situ Assembling of Gold/Silver Nanoparticles. ChemistryOpen 2019, 8, 1369–1374.
- Kohler, J.M.; Wagner, J.; Albert, J. Formation of isolated and clustered Au nanoparticles in the presence of polyelectrolyte molecules using a flow-through Si chip reactor. J. Mater. Chem. 2005, 15, 1924–1930.
- Mahmoud, M.A.; El-Sayed, M.A. Gold nanoframes: Very high surface plasmon fields and excellent near-infrared sensors. JACS 2010, 132, 12704–12710.
- Cheng, S.; Su, H.; Wang, Y.; Wu, W.; Zeng, J. Size-controlled synthesis of platinum-copper hierarchical trigonal bipyramid nanoframes. Angew. Chem. Int. Ed. Engl. 2015, 54, 108–113.
- Kluitmann, J.J.; Köhler, J.M. Tuning the morphology of bimetallic gold-platinum nanorods in a microflow synthesis. Colloids Surf. A Physicochem. Eng. Asp. 2021, 626, 127085.