The conversion of solar energy and water to hydrogen via semiconductor photocatalysts is one of the efficient strategies to mitigate the energy and environmental crisis. Conjugated polymeric photocatalysts have advantages over their inorganic counterparts. Their molecular structures, band structures, and electronic properties are easily tunable through molecular engineering to extend their spectral response ranges, improve their quantum efficiencies, and enhance their hydrogen evolution rates. In particular, covalent triazine-based frameworks (CTFs) present a strong potential for solar-driven hydrogen generation due to their large continuous π-conjugated structure, high thermal and chemical stability, and efficient charge transfer and separation capability.
1. Introduction
The ability to effectively harness and store renewable energy in chemical form has been widely recognized as a promising and sustainable strategy to meet future worldwide energy demands. Solar energy is by far one of the most widely distributed renewable primary energy sources. As a secondary energy source, hydrogen energy is generally considered an ideal energy carrier due to its high energy density, zero emissions, storability, and transportability. Therefore, converting solar energy into hydrogen in a single step is of great significance for the efficient and clean utilization of solar energy, reducing carbon emissions and environmental pollution [1][2].
2. Optimization of CTFs for Photocatalytic Hydrogen Generation
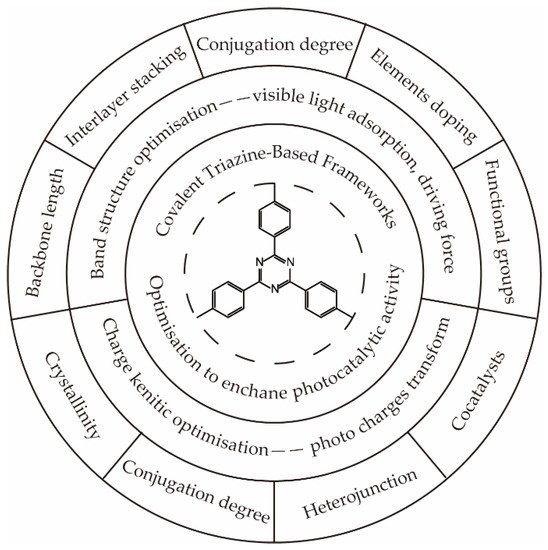
Although some efforts have been devoted to exploring efficient CTFs for solar-driven hydrogen generation, most of them focus on screening the block or loading cocatalysts to achieve high activity, ignoring the investigation of the mechanism. The application of highly conjugated polymer CTFs in photocatalysis was first predicted by first-principle calculations in 2015 [3]. According to the modeling results, covalent triazine-based frameworks (CTFs) had a suitable band structure that could effectively be excited by UV and visible light. The generated photoelectrons generated at −0.5 eV–−1 eV (vs. NHE) were sufficient to drive the proton reduction reaction to generate hydrogen. The energy of photogenerated holes was predicted between +2 eV and +1.2 eV (vs. NHE), which were able to oxidize water to form molecular oxygen. To improve the efficiency of photocatalytic hydrogen generation and investigate the relationship between the catalyst structure and performance, as presented in
1. Introduction
The ability to effectively harness and store renewable energy in chemical form has been widely recognized as a promising and sustainable strategy to meet future worldwide energy demands. Solar energy is by far one of the most widely distributed renewable primary energy sources. As a secondary energy source, hydrogen energy is generally considered an ideal energy carrier due to its high energy density, zero emissions, storability, and transportability. Therefore, converting solar energy into hydrogen in a single step is of great significance for the efficient and clean utilization of solar energy, reducing carbon emissions and environmental pollution [1,2].
2. Optimization of CTFs for Photocatalytic Hydrogen Generation
Although some efforts have been devoted to exploring efficient CTFs for solar-driven hydrogen generation, most of them focus on screening the block or loading cocatalysts to achieve high activity, ignoring the investigation of the mechanism. The application of highly conjugated polymer CTFs in photocatalysis was first predicted by first-principle calculations in 2015 [34]. According to the modeling results, covalent triazine-based frameworks (CTFs) had a suitable band structure that could effectively be excited by UV and visible light. The generated photoelectrons generated at −0.5 eV–−1 eV (vs. NHE) were sufficient to drive the proton reduction reaction to generate hydrogen. The energy of photogenerated holes was predicted between +2 eV and +1.2 eV (vs. NHE), which were able to oxidize water to form molecular oxygen. To improve the efficiency of photocatalytic hydrogen generation and investigate the relationship between the catalyst structure and performance, as presented in Figure 1, a great number of modification strategies have been performed to optimize the band structure [4][5][6][7][8][9][10][11][12] and charge dynamic process [13][14][15][16][17][18][19][20][21][22], thus broadening the visible light absorption, enhancing the driving force of hydrogen generation, and improving the transport and separation efficiency of photocharges.
, a great number of modification strategies have been performed to optimize the band structure [12,14,20,35,36,37,38,39,40] and charge dynamic process [10,15,41,42,43,44,45,46,47,48], thus broadening the visible light absorption, enhancing the driving force of hydrogen generation, and improving the transport and separation efficiency of photocharges.
Figure 1. Functionalized optimization of the covalent triazine-based framework.
2.1. Optimization of Band Structure to Broaden the Visible Light Absorption and Enhance the Driving Force for Hydrogen Generation
The band structure of photocatalysts determines the light response range and the energy of photogenerated charges. A smaller bandgap leads to a higher solar utilization rate. On the contrary, hydrogen generation is an endothermic reaction; thus, the energy of photogenerated charges must meet both the thermodynamic requirements and the overpotential for hydrogen generation. Therefore, the bandgap optimization of photocatalysts is necessary to strike a balance between light response and sufficient driving force.
2.1.1. Optimize the Length of the Backbone
The distribution of aromatic and triazine motifs in CTFs determines the energy level of both the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Different quantities of benzene units are inserted between two triazine motifs to increase the length of the backbones. CTFs with different lengths of backbones were synthesized by dehydration polycondensation [23], Suzuki coupling [24] and nitrile trimerization [24]. The experimental results illustrated that the longer the backbone, the narrower the bandgap of CTFs, and the higher the efficiency of photonic utilization. More significantly, the number of aromatic spacers greatly impacted the energy of HOMO while maintaining the position of LUMO; thus, the optimized materials kept a similar driving force for hydrogen generation. However, with an increase in the aromatic proportion of the materials, the hydrophilicity deteriorated. Therefore, the mass transfer between the catalyst and water molecule became worse when increasing the length of the backbone by adding aromatic spacers. When two triazine units were spaced by two benzene motifs, the highest hydrogen generation rate was observed on the photocatalysts prepared by all three methods. The reported hydrogen generation rate was 7.4 µmol h Functionalized optimization of the covalent triazine-based framework.
2.1. Optimization of Band Structure to Broaden the Visible Light Absorption and Enhance the Driving Force for Hydrogen Generation
The band structure of photocatalysts determines the light response range and the energy of photogenerated charges. A smaller bandgap leads to a higher solar utilization rate. On the contrary, hydrogen generation is an endothermic reaction; thus, the energy of photogenerated charges must meet both the thermodynamic requirements and the overpotential for hydrogen generation. Therefore, the bandgap optimization of photocatalysts is necessary to strike a balance between light response and sufficient driving force.
2.1.1. Optimize the Length of the Backbone
The distribution of aromatic and triazine motifs in CTFs determines the energy level of both the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Different quantities of benzene units are inserted between two triazine motifs to increase the length of the backbones. CTFs with different lengths of backbones were synthesized by dehydration polycondensation [23], Suzuki coupling [29] and nitrile trimerization [29]. The experimental results illustrated that the longer the backbone, the narrower the bandgap of CTFs, and the higher the efficiency of photonic utilization. More significantly, the number of aromatic spacers greatly impacted the energy of HOMO while maintaining the position of LUMO; thus, the optimized materials kept a similar driving force for hydrogen generation. However, with an increase in the aromatic proportion of the materials, the hydrophilicity deteriorated. Therefore, the mass transfer between the catalyst and water molecule became worse when increasing the length of the backbone by adding aromatic spacers. When two triazine units were spaced by two benzene motifs, the highest hydrogen generation rate was observed on the photocatalysts prepared by all three methods. The reported hydrogen generation rate was 7.4 µmol h −1
(>420 nm) for the catalyst prepared by polycondensation, 118.31 µmol h
−1
(>420 nm) for the catalyst prepared by Suzuki coupling, and 132.15 µmol h
−1
(>420 nm) for the catalyst prepared by nitrile trimerization. The activity difference among these samples was likely due to the different crystal structures and sp
2.1.2. Control the Interlayer Stacking
CTFs are 2D-layered conjugated materials. By controlling the stacking between layers, functional groups can have different interlayer interactions resulting in various distributions of electron clouds. CTF-0 with eclipsed AA stacking and staggered AB stacking were synthesized by ionothermal synthesis and microwave-assisted synthesis, respectively [4]. In the eclipsed stacked CTF-0, triazine corresponded to triazine, and benzene corresponded to triazine between two layers. Conversely, in the staggered stacked CTF-0, triazine corresponded to benzene between odd and even layers. Therefore, the interlayer n⟶π transition was possible in the staggered stacked CTFs. Both theoretical and experimental results indicated that interlayer excitation resulted in a more negative conduction band. Thus, the excited photoelectrons held higher energy to drive proton reduction. Therefore, the photocatalytic hydrogen evolution for the sample with AB stacking was 100 µmol h
2.1.2. Control the Interlayer Stacking
CTFs are 2D-layered conjugated materials. By controlling the stacking between layers, functional groups can have different interlayer interactions resulting in various distributions of electron clouds. CTF-0 with eclipsed AA stacking and staggered AB stacking were synthesized by ionothermal synthesis and microwave-assisted synthesis, respectively [12]. In the eclipsed stacked CTF-0, triazine corresponded to triazine, and benzene corresponded to triazine between two layers. Conversely, in the staggered stacked CTF-0, triazine corresponded to benzene between odd and even layers. Therefore, the interlayer n⟶π transition was possible in the staggered stacked CTFs. Both theoretical and experimental results indicated that interlayer excitation resulted in a more negative conduction band. Thus, the excited photoelectrons held higher energy to drive proton reduction. Therefore, the photocatalytic hydrogen evolution for the sample with AB stacking was 100 µmol h −1
(>420 nm), which was much higher than samples with AA stacking of 9 µmol h
−1 (>420 nm) [4]. A redox exfoliation process was utilized to obtain ultrathin crystalline CTF. The overall thinness of r-CTF was only 1.2 nm, indicating 3–4 sheets in a piece of crystallized CTF, resulting in a photocatalytic hydrogen generation rate of 500 µmol h (>420 nm) [12]. A redox exfoliation process was utilized to obtain ultrathin crystalline CTF. The overall thinness of r-CTF was only 1.2 nm, indicating 3–4 sheets in a piece of crystallized CTF, resulting in a photocatalytic hydrogen generation rate of 500 µmol h
2.1.3. Control the Degree of Polymerization
The degree of polymerization affects the number of terminal groups and the distance between layers. By controlling the reaction temperature, the ratio between ZnCl2 and monomer, and the polymerization time, the degree of polymerization was tuned via ionothermal synthesis [6]. The experimental results showed that the lower the degree of polymerization, the smaller the interlayer spacing, and the higher the hydrophilicity. However, the bandgap due to the π⟶π* transition widened, thus reducing light absorption efficiently. The benchmark photocatalytic activity of 10.8 (±2.8) µmol h
2.1.3. Control the Degree of Polymerization
The degree of polymerization affects the number of terminal groups and the distance between layers. By controlling the reaction temperature, the ratio between ZnCl2 and monomer, and the polymerization time, the degree of polymerization was tuned via ionothermal synthesis [20]. The experimental results showed that the lower the degree of polymerization, the smaller the interlayer spacing, and the higher the hydrophilicity. However, the bandgap due to the π⟶π* transition widened, thus reducing light absorption efficiently. The benchmark photocatalytic activity of 10.8 (±2.8) µmol h −1 (solar simulator) was achieved via an almost linear oligomer PTO-300-15 with a molecular weight of 2000–3000 and an apparent quantum efficiency of 5.5 ± 1.1% (at 400 ± 20 nm).
2.1.4. Element Doping
Heteroatom doping effectively changes the electron distribution of polymeric materials. In general, the doping of O [11], S [7], P [8], and other elements mainly contributed to HOMO electron distribution and changed the position of the valence band. Thus, it is possible to narrow the bandgap while maintaining the position of the conduction band to supply enough driving force for hydrogen generation. To dope sulfur into CTF-1, the polymerized CTF-1 was ground with sulfur powder and calcined at 250 °C for 1 h under a nitrogen atmosphere. The bandgap was sufficiently narrowed. Compared with the undoped sample, the photocatalytic activity for hydrogen generation was enhanced 4 times [7]. When replacing N atoms in CTF-1 with P atoms, a narrower bandgap was obtained as well, while the photocatalytic activity increased 5 times compared to the bare sample [8]. However, elemental doping may destroy the conjugated structure of the material and cause a great deviation in LUMO position as well. For the oxygen doping materials in particular, one study reported hydrogen had an evolution rate of only 0.5 µmol h (solar simulator) was achieved via an almost linear oligomer PTO-300-15 with a molecular weight of 2000–3000 and an apparent quantum efficiency of 5.5 ± 1.1% (at 400 ± 20 nm).
2.1.4. Element Doping
Heteroatom doping effectively changes the electron distribution of polymeric materials. In general, the doping of O [39], S [35], P [36], and other elements mainly contributed to HOMO electron distribution and changed the position of the valence band. Thus, it is possible to narrow the bandgap while maintaining the position of the conduction band to supply enough driving force for hydrogen generation. To dope sulfur into CTF-1, the polymerized CTF-1 was ground with sulfur powder and calcined at 250 °C for 1 h under a nitrogen atmosphere. The bandgap was sufficiently narrowed. Compared with the undoped sample, the photocatalytic activity for hydrogen generation was enhanced 4 times [35]. When replacing N atoms in CTF-1 with P atoms, a narrower bandgap was obtained as well, while the photocatalytic activity increased 5 times compared to the bare sample [36]. However, elemental doping may destroy the conjugated structure of the material and cause a great deviation in LUMO position as well. For the oxygen doping materials in particular, one study reported hydrogen had an evolution rate of only 0.5 µmol h −1 (>420 nm), which was much lower than the bare sample [11]. This could partially be explained by the halogen atoms doped samples. When treating CTF-1 with HCl, Cl atoms were successfully doped in the position between the C-N bond, showing much lower activity than the undoped sample [10]. On the contrary, when treated CTF-1 by NH4Cl, Cl atoms were only able to substitute H atoms in benzene motifs. The photocatalytic activity of the Cl-doped sample increased 7.1 times compared to the unmodified sample, and high quantum efficiency of 10.31% was achieved under 420 nm light irradiation [9]. By comparing the band structures of two Cl-doped samples, the conduction band slightly shifted to more negative as well when replacing H in benzene, although the overall bandgap was narrower than the unmodified sample, which thus gave more driving force for proton reduction while increasing the light absorption range. Fe (>420 nm), which was much lower than the bare sample [39]. This could partially be explained by the halogen atoms doped samples. When treating CTF-1 with HCl, Cl atoms were successfully doped in the position between the C-N bond, showing much lower activity than the undoped sample [38]. On the contrary, when treated CTF-1 by NH4Cl, Cl atoms were only able to substitute H atoms in benzene motifs. The photocatalytic activity of the Cl-doped sample increased 7.1 times compared to the unmodified sample, and high quantum efficiency of 10.31% was achieved under 420 nm light irradiation [37]. By comparing the band structures of two Cl-doped samples, the conduction band slightly shifted to more negative as well when replacing H in benzene, although the overall bandgap was narrower than the unmodified sample, which thus gave more driving force for proton reduction while increasing the light absorption range. Fe 3+
ions were also claimed to be doped into the CTF molecule structure. With the increase in Fe amount, the valence band position shifted more negatively, resulting in a 30 times improvement in the hydrogen generation rate than the bare sample of 30 µmol h
−1 (>420 nm) [27]. Protonating CTFs by acid was a strategy to enhance hydrophilicity. The protonated CTF-1 presented a contact angle of 38.4°, which was only one-third of the bare sample, resulting in ca. 10 times enhancement of the hydrogen evolution rate [28].
2.1.5. Functional Group Substitution
According to theoretical calculations, in a typical CTF molecule, HOMO is determined by the triazine motif, and LUMO is associated with the benzene unit. Therefore, proton reduction occurred on benzene sites [3]. However, due to the hydrogen bond between water and benzene, it is not conducive to dissociating water molecules. Therefore, the backbones of CTFs were functionalized to relocalize the electron distribution and optimize the active sites. Carbonyl carbazole, dibenzothiophene, and dibenzofuran were utilized as the aldehyde monomer for the dehydration polycondensation, thus introducing the above functional groups into the polymer backbone. The highest photocatalytic activity of 538 µmol h (>420 nm) [50]. Protonating CTFs by acid was a strategy to enhance hydrophilicity. The protonated CTF-1 presented a contact angle of 38.4°, which was only one-third of the bare sample, resulting in ca. 10 times enhancement of the hydrogen evolution rate [51].
2.1.5. Functional Group Substitution
According to theoretical calculations, in a typical CTF molecule, HOMO is determined by the triazine motif, and LUMO is associated with the benzene unit. Therefore, proton reduction occurred on benzene sites [34]. However, due to the hydrogen bond between water and benzene, it is not conducive to dissociating water molecules. Therefore, the backbones of CTFs were functionalized to relocalize the electron distribution and optimize the active sites. Carbonyl carbazole, dibenzothiophene, and dibenzofuran were utilized as the aldehyde monomer for the dehydration polycondensation, thus introducing the above functional groups into the polymer backbone. The highest photocatalytic activity of 538 µmol h −1 (>420 nm) for hydrogen generation, and the quantum efficiency of 4.07% (at 420 nm) was over that of the carbazole-functionalized sample CTF-N [12]. The enhancement of the activity was due to the efficient charge transfer from the introduced functional groups to the triazine motifs and the narrowed bandgap. Furthermore, the active sites for proton reduction shifted from benzene to triazine. The lone-pair electrons in triazine were sufficient for the dissociation of water molecules, thus further enhancing the photocatalytic activity. Introducing two functional groups such as pyrazole and benzothiadiazole to change the activation sites for water oxidation and proton reduction, respectively, resulted in much higher hydrophilicity and better interaction between water and photocatalysts [4][29]. However, the backbone of the polymer became more distorted when introducing more functional groups, resulting in a low efficiency for electron transport. The hydrogen generation rate was relatively low at 50 µmol h (>420 nm) for hydrogen generation, and the quantum efficiency of 4.07% (at 420 nm) was over that of the carbazole-functionalized sample CTF-N [40]. The enhancement of the activity was due to the efficient charge transfer from the introduced functional groups to the triazine motifs and the narrowed bandgap. Furthermore, the active sites for proton reduction shifted from benzene to triazine. The lone-pair electrons in triazine were sufficient for the dissociation of water molecules, thus further enhancing the photocatalytic activity. Introducing two functional groups such as pyrazole and benzothiadiazole to change the activation sites for water oxidation and proton reduction, respectively, resulted in much higher hydrophilicity and better interaction between water and photocatalysts [12,13]. However, the backbone of the polymer became more distorted when introducing more functional groups, resulting in a low efficiency for electron transport. The hydrogen generation rate was relatively low at 50 µmol h −1
(>420 nm), and the quantum efficiency was 3.58% at 420 ± 20 nm. Thiophene and benzothiadiazole were introduced into the backbone of CTF via the cyano trimerization method. The HOMO level shifted greatly to narrower the bandgap while maintaining the LUMO level to enhance the light absorption. A hydrogen evolution rate of 112 µmol h
−1 (>420 nm) was observed [30].
2.2. Control of Charge Dynamic to Enhance the Electron Transfer and Separation
A typical photocatalytic process can be described in three steps: (1) photon absorption by a semiconductor to generate excited charge carriers, (2) diffusion of photocharges to semiconductor surface, and (3) interfacial charge transfer between active sites, followed by the transfer to reactants. However, the excited photoelectrons and holes could instead recombine to determine the lifetime of the charge carriers. Furthermore, the lifetime of charge carriers directly impacted the diffusion length of photocharges. Therefore, the optimization of the charge dynamic by controlling the structure of photocatalysts was greatly significant.
2.2.1. Enhance the Degree of Crystallinity
Both crystallinity and crystallite dimensions affect the efficiency of charge transfer and separation. Low crystallinity is due to the low stereoregularity and more defects in the molecule structure, both of which may result in the formation of recombination centers to obstruct charge transfer. On the contrary, high crystallinity lead to a large crystallite size. With an increase in crystallinity degree, the electrical conductivity raise, but the ionic conductance decrease. Thus, the diffusion of dissociated water species are limited [31]. Samples synthesized by triazine trimerization via solution polymerization had very poor crystallinity. The photocatalytic activity for hydrogen generation was very low, around 8 µmol h (>420 nm) was observed [52].
2.2. Control of Charge Dynamic to Enhance the Electron Transfer and Separation
A typical photocatalytic process can be described in three steps: (1) photon absorption by a semiconductor to generate excited charge carriers, (2) diffusion of photocharges to semiconductor surface, and (3) interfacial charge transfer between active sites, followed by the transfer to reactants. However, the excited photoelectrons and holes could instead recombine to determine the lifetime of the charge carriers. Furthermore, the lifetime of charge carriers directly impacted the diffusion length of photocharges. Therefore, the optimization of the charge dynamic by controlling the structure of photocatalysts was greatly significant.
2.2.1. Enhance the Degree of Crystallinity
Both crystallinity and crystallite dimensions affect the efficiency of charge transfer and separation. Low crystallinity is due to the low stereoregularity and more defects in the molecule structure, both of which may result in the formation of recombination centers to obstruct charge transfer. On the contrary, high crystallinity lead to a large crystallite size. With an increase in crystallinity degree, the electrical conductivity raise, but the ionic conductance decrease. Thus, the diffusion of dissociated water species are limited [53]. Samples synthesized by triazine trimerization via solution polymerization had very poor crystallinity. The photocatalytic activity for hydrogen generation was very low, around 8 µmol h −1 (>420 nm) [32]. To mix the above sample with ZnCl2 and calcined under vacuum at 400 °C for 2.5 to 30 min, the crystallinity obviously increased. Among all treated samples, the material calcined 10 min presented the highest hydrogen generation rate of 26.8 µmol h (>420 nm) [25]. To mix the above sample with ZnCl2 and calcined under vacuum at 400 °C for 2.5 to 30 min, the crystallinity obviously increased. Among all treated samples, the material calcined 10 min presented the highest hydrogen generation rate of 26.8 µmol h −1
(>420 nm). More importantly, the quantum efficiency increased from 2.4% of the uncalcined sample to 6.4% of the treated sample. When calcining the sample for more than 10 min, the crystallinity further increased, while the photocatalytic activity decreased, probably attributed to the lower efficiency for the ionic conductance and the carbonization of the materials to block the light adsorption. By using a different base catalyst such as K2CO3, KOH, EtOK, and
tBuOK, CTF-1s with different crystallinity were synthesized via Michael addition. The photocatalytic activity was positively correlated with the crystallinity of the synthesized CTF-1s [14]. The sample synthesized over BuOK, CTF-1s with different crystallinity were synthesized via Michael addition. The photocatalytic activity was positively correlated with the crystallinity of the synthesized CTF-1s [15]. The sample synthesized over t
BuOK presented the highest hydrogen generation rate of 335 µmol h
−1
(>420 nm) with a quantum efficiency of 7.4% at 420 nm. With the increase in the crystallinity, the lifetime of photocharges increased from 214.5 ps to 2630 ps, while the contact angles of water droplets on the surface decreased from 65.5° to ca. 5°. More importantly, when using Pt nanoparticles and NiPx as the cocatalysts for proton reduction and water oxidation, respectively, efficient overall water splitting was observed with a hydrogen generation rate of 25.4 µmol h
−1 [14]. A NaCl-KCl-ZnCl2 eutectic salt system reduced the polymerization temperature during ionothermal synthesis from 400 °C to 200 °C to avoid the carbonization process. Thus, a high crystallized sample was observed compared to that synthesized under high temperatures. A high degree of polymerization was also confirmed by FTIR. A hydrogen evolution rate of 60 µmol h [15]. A NaCl-KCl-ZnCl2 eutectic salt system reduced the polymerization temperature during ionothermal synthesis from 400 °C to 200 °C to avoid the carbonization process. Thus, a high crystallized sample was observed compared to that synthesized under high temperatures. A high degree of polymerization was also confirmed by FTIR. A hydrogen evolution rate of 60 µmol h −1 was achieved, which was 3 times higher than the sample with poor crystallinity synthesized by ZnCl2 only at 400 °C [33].
2.2.2. Control the Degree of Conjugation
For CTFs that are layered polymers, the degree of conjugation represents the distortion of the polymeric material that originated from sp
was achieved, which was 3 times higher than the sample with poor crystallinity synthesized by ZnCl2 only at 400 °C [54].
2.2.2. Control the Degree of Conjugation
For CTFs that are layered polymers, the degree of conjugation represents the distortion of the polymeric material that originated from sp
3
defects during synthesis. The higher the stereospecificity, the more efficient the interlayer charge transition and the within-layer charge transfer. The degree of conjugation of CTFs was able to be characterized by Raman spectra by calculating the ratio of sp
2
3
. By controlling the microwave power, CTF-1s with different degrees of conjugation were synthesized by microwave-assisted synthesis. With the increasing conjugation degree, the efficiency of charge transfer greatly enhanced, resulting in a high hydrogen generation rate of 265 µmol h
−1 with a quantum efficiency of 6.0% at 420 nm.
2.2.3. Heterojunction Construction
A heterojunction is an interface region that results from the contact of two different semiconductors. Heterojunction construction allows photogenerated electrons and holes to be stored on different semiconductors, respectively, so that the hydrogen evolution and water oxidation reactions are able to be carried out on two counterparts, which promotes the transfer and utilization of photogenerated charges. CTF-1 was combined with CdS to form a type II heterojunction. According to electrochemical impedance spectroscopy and photoluminescence spectroscopy, the charge transfer of the composite was much stronger than that of two single materials, CdS and CTF-1. Using lactic acid as the sacrificial agent, the hydrogen evolution rate was 243 µmol h
with a quantum efficiency of 6.0% at 420 nm.
2.2.3. Heterojunction Construction
A heterojunction is an interface region that results from the contact of two different semiconductors. Heterojunction construction allows photogenerated electrons and holes to be stored on different semiconductors, respectively, so that the hydrogen evolution and water oxidation reactions are able to be carried out on two counterparts, which promotes the transfer and utilization of photogenerated charges. CTF-1 was combined with CdS to form a type II heterojunction. According to electrochemical impedance spectroscopy and photoluminescence spectroscopy, the charge transfer of the composite was much stronger than that of two single materials, CdS and CTF-1. Using lactic acid as the sacrificial agent, the hydrogen evolution rate was 243 µmol h
−1 (>420 nm), which was three times higher than that achieved by CdS nanoparticles [10]. Furthermore, as both conduction and valence band positions of CTF-1 were more positive than that of CdS, photogenerated electrons were directed to the conduction band of CdS, while photoholes transferred to the valence band of CTF-1, which effectively avoid the oxidative corrosion of CdS by photoholes. Therefore, the stability of the composite was much higher than CdS resulting in a 36 h continuous hydrogen generation without activity decay [15]. Similar phenomena were observed over the composite of MoS2 and CTF-1 with a quantum efficiency of ca. 7% at 420 nm [17].
Thiophene and benzothiadiazole were introduced at various positions of the backbones of CTFs. Thus, a nanocomposite was formed as a type II heterojunction. Photoelectrons were concentrated in the benzothiadiazole side as the different HOMO and LUMO structures of two parts of the backbones. Therefore, the density of photoelectrons was enhanced. Meanwhile, as the polymer was based on benzene and triazine motifs, the charge transfer efficiency between the two parts was much higher than the charge transfer in conjugated polymers and metal-based compounds such as CdS, MoS2, etc. Therefore, such an amazing structure allowed it to present a hydrogen generation rate of 330 µmol h
(>420 nm), which was three times higher than that achieved by CdS nanoparticles [38]. Furthermore, as both conduction and valence band positions of CTF-1 were more positive than that of CdS, photogenerated electrons were directed to the conduction band of CdS, while photoholes transferred to the valence band of CTF-1, which effectively avoid the oxidative corrosion of CdS by photoholes. Therefore, the stability of the composite was much higher than CdS resulting in a 36 h continuous hydrogen generation without activity decay [41]. Similar phenomena were observed over the composite of MoS2 and CTF-1 with a quantum efficiency of ca. 7% at 420 nm [43].
Thiophene and benzothiadiazole were introduced at various positions of the backbones of CTFs. Thus, a nanocomposite was formed as a type II heterojunction. Photoelectrons were concentrated in the benzothiadiazole side as the different HOMO and LUMO structures of two parts of the backbones. Therefore, the density of photoelectrons was enhanced. Meanwhile, as the polymer was based on benzene and triazine motifs, the charge transfer efficiency between the two parts was much higher than the charge transfer in conjugated polymers and metal-based compounds such as CdS, MoS2, etc. Therefore, such an amazing structure allowed it to present a hydrogen generation rate of 330 µmol h
−1 (>420 nm) and a quantum efficiency of 7.3% at 420 nm [18]. When using N-ethylcarbazole to replace thiophene in the nanocomposite, the energy band difference between electron donor and acceptor was increased. Therefore, the hydrogen generation rate was further enhanced to 966 µmol h (>420 nm) and a quantum efficiency of 7.3% at 420 nm [44]. When using N-ethylcarbazole to replace thiophene in the nanocomposite, the energy band difference between electron donor and acceptor was increased. Therefore, the hydrogen generation rate was further enhanced to 966 µmol h −1 (>420 nm), and the quantum efficiency was 22.8% [19].
2.2.4. Cocatalyst Deposition
Noble metals are typically considered cocatalysts for hydrogen generation. Among them, the free Gibbs energy for hydrogen dissociation is close to 0 for Pt, Pd, and Rh, which is much smaller than that on the surface of Au, Ag, Cu, and Ni. Therefore, Pt, Pd, and Rh are loaded. Meanwhile, the work functions of such noble metals are between the conduction and valence bands of CTFs. Therefore, a Schottky junction is able to be formed to direct photoelectrons to the cocatalyst. The majority of cocatalyst utilized in the previously reviewed measurements were Pt nanoparticles. There were mainly three deposition paths, and all start from chloroplatinic acid: (1) impregnation of chloroplatinic acid with CTFs and calcinated in the air to decomposed; (2) utilizing sodium borohydride to reduce platinum in solution; (3) photodeposited platinum in situ by photogenerated electrons. All three routes were able to anchorite Pt nanoparticles on the surface of CTFs. However, due to the high conjugated surface of CTFs, it was difficult to form chemical bonds between Pt nanoparticles and CTFs. Thus, the efficiency of electron transfer was limited. In order to enhance the metal–semiconductor contact and improve the electron transfer, black phosphorus served as a bridge to connect the Pt nanoparticles and CTFs by forming N-P-Pt chemical bonds. The hydrogen generation rate over Pt/P/CTF-1 was 5 times higher than that of Pt/CTF-1 [20]. However, black phosphorus had a band structure, thus forming a type I heterojunction when deposited on CTF-1. Therefore, both photogenerated electrons and holes would transfer to black phosphorus, resulting in the recombination [21][34]. To overcome the drawbacks of the band structure of black phosphorus, one of the carbon atoms in the benzene motif was replaced by a nitrogen atom to form a bipyridine-like structure with triazine. The bidentate ligands were able to interact with Pt and Pd chlorides from the chemical bond between CTFs and noble metal active centers. The hydrogen generation rate of Pd decorated nitrogen-doped CTF reached 106 µmol h (>420 nm), and the quantum efficiency was 22.8% [45].
2.2.4. Cocatalyst Deposition
Noble metals are typically considered cocatalysts for hydrogen generation. Among them, the free Gibbs energy for hydrogen dissociation is close to 0 for Pt, Pd, and Rh, which is much smaller than that on the surface of Au, Ag, Cu, and Ni. Therefore, Pt, Pd, and Rh are loaded. Meanwhile, the work functions of such noble metals are between the conduction and valence bands of CTFs. Therefore, a Schottky junction is able to be formed to direct photoelectrons to the cocatalyst. The majority of cocatalyst utilized in the previously reviewed measurements were Pt nanoparticles. There were mainly three deposition paths, and all start from chloroplatinic acid: (1) impregnation of chloroplatinic acid with CTFs and calcinated in the air to decomposed; (2) utilizing sodium borohydride to reduce platinum in solution; (3) photodeposited platinum in situ by photogenerated electrons. All three routes were able to anchorite Pt nanoparticles on the surface of CTFs. However, due to the high conjugated surface of CTFs, it was difficult to form chemical bonds between Pt nanoparticles and CTFs. Thus, the efficiency of electron transfer was limited. In order to enhance the metal–semiconductor contact and improve the electron transfer, black phosphorus served as a bridge to connect the Pt nanoparticles and CTFs by forming N-P-Pt chemical bonds. The hydrogen generation rate over Pt/P/CTF-1 was 5 times higher than that of Pt/CTF-1 [46]. However, black phosphorus had a band structure, thus forming a type I heterojunction when deposited on CTF-1. Therefore, both photogenerated electrons and holes would transfer to black phosphorus, resulting in the recombination [47,55]. To overcome the drawbacks of the band structure of black phosphorus, one of the carbon atoms in the benzene motif was replaced by a nitrogen atom to form a bipyridine-like structure with triazine. The bidentate ligands were able to interact with Pt and Pd chlorides from the chemical bond between CTFs and noble metal active centers. The hydrogen generation rate of Pd decorated nitrogen-doped CTF reached 106 µmol h −1 (>420 nm) [22]. Another strategy was to directly utilize Ni2P as the cocatalyst to get a better interaction for charge transfer. However, as the higher driving force for the hydrogen evolution over Ni sites the hydrogen evolution over Ni2P/CTF was similar to Pt/CTF [35]. rGO was also investigated as the cocatalyst to enhance the charge separation, the hydrogen evolution was of 20–30 µmol h (>420 nm) [48]. Another strategy was to directly utilize Ni2P as the cocatalyst to get a better interaction for charge transfer. However, as the higher driving force for the hydrogen evolution over Ni sites the hydrogen evolution over Ni2P/CTF was similar to Pt/CTF [56]. rGO was also investigated as the cocatalyst to enhance the charge separation, the hydrogen evolution was of 20–30 µmol h −1 (>420 nm), which was more than 10 times higher than the bare sample under the identical conditions [36][37]. (>420 nm), which was more than 10 times higher than the bare sample under the identical conditions [57,58].