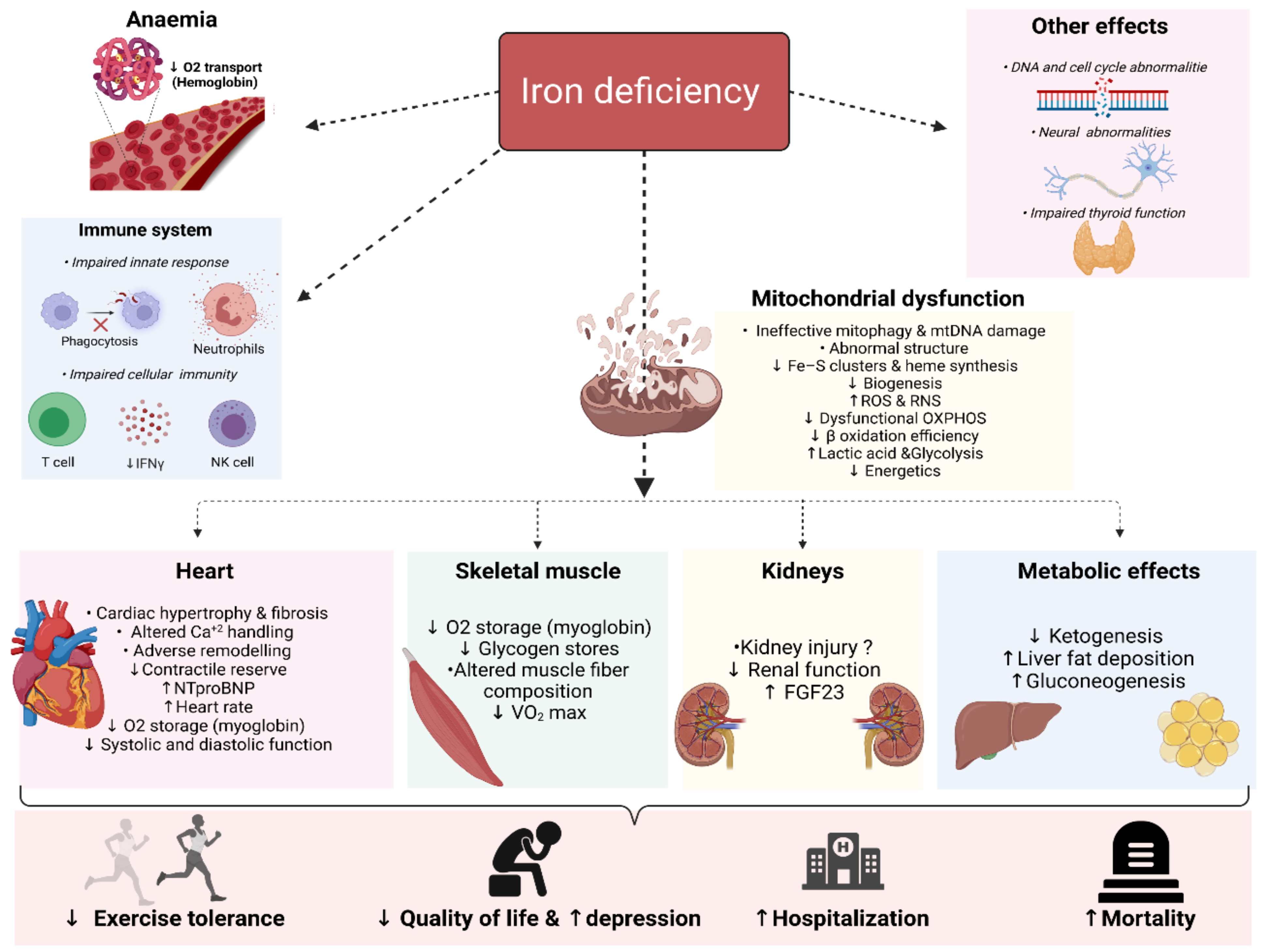
Iron is an essential micronutrient for a myriad of physiological processes in the body beyond erythropoiesis. Iron deficiency (ID) is a common comorbidity in patients with heart failure (HF), with a prevalence reaching up to 59% even in non-anaemic patients. ID impairs exercise capacity, reduces the quality of life, increases hospitalisation rate and mortality risk regardless of anaemia. Intravenously correcting ID has emerged as a promising treatment in HF as it has been shown to alleviate symptoms, improve quality of life and exercise capacity and reduce hospitalisations. However, the pathophysiology of ID in HF remains poorly characterised. Recognition of ID in HF triggered more research with the aim to explain how correcting ID improves HF status as well as the underlying causes of ID in the first place. In the past few years, significant progress has been made in understanding iron homeostasis by characterising the role of the iron-regulating hormone hepcidin, the effects of ID on skeletal and cardiac myocytes, kidneys and the immune system. In this review, we summarise the current knowledge and recent advances in the deleterious systemic and cellular consequences of ID in HF.
- iron deficiency
- iron metabolism
- heart failure
- pathophysiology
1. Introduction
1.1. Physiologic Roles and Regulation of Iron
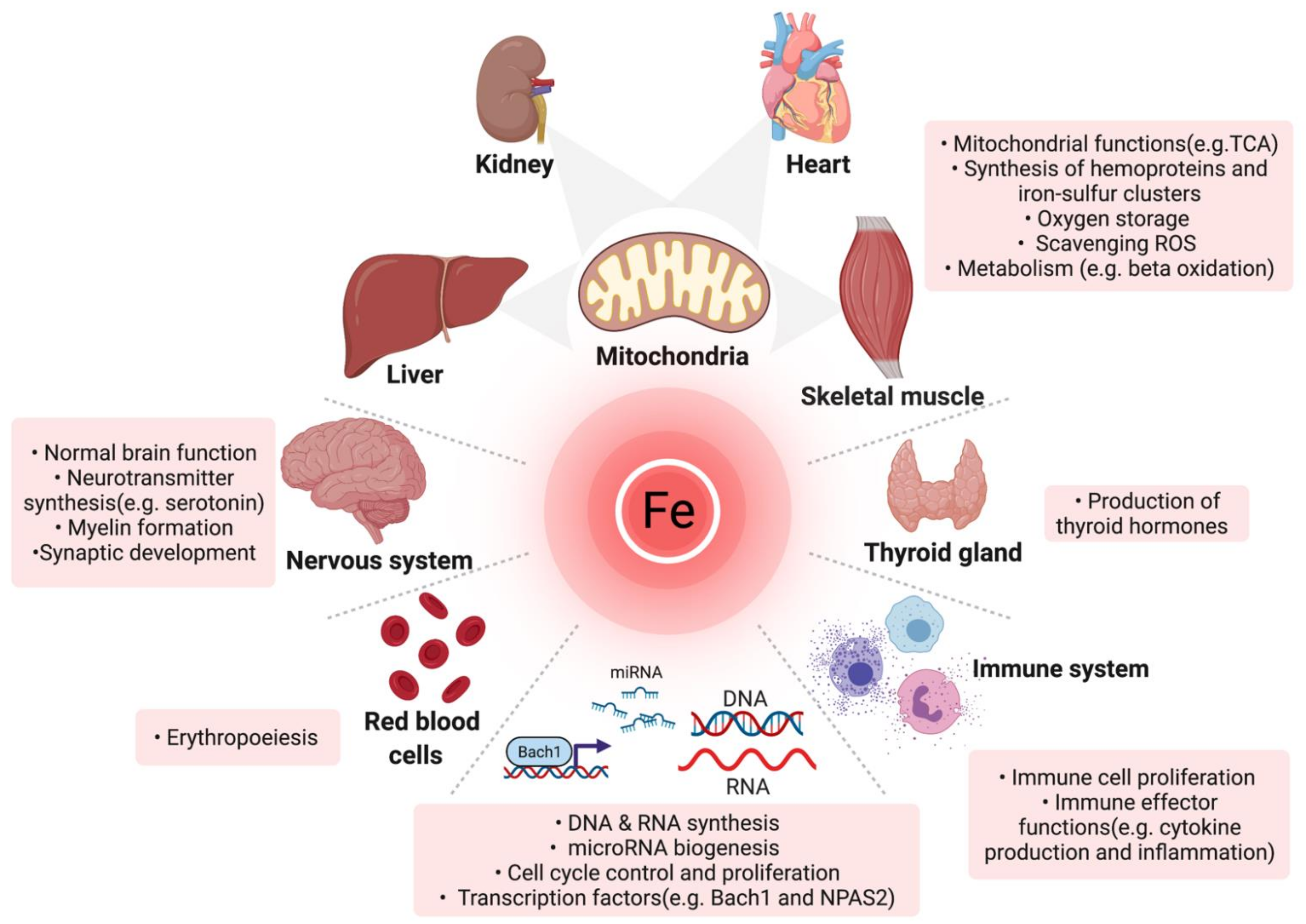
| Function | Protein |
|---|---|
| Oxygen transport | Hemoglobin |
| Oxygen storage | Myoglobin |
| Lipid and cholesterol biosynthesis | NADPH-cytochrome P450 reductase, fatty acid desaturases, cytochrome P-450 subfamily 51 and Cytochrome P450 Family 7 Subfamily A Member 1 |
| Oxygen sensing and regulation of hypoxia | Hypoxia-inducible factor prolyl hydroxylases |
| Synthesis catecholamines and neurotransmitters | Tryptophan hydroxylase, tyrosine hydroxylase, monoamine oxidase and aldehyde oxidase |
| Host defence, inflammation and production of nitric oxide | Myeloperoxidase, NADPH oxidase, indoleamine 2,3- dioxygenase, nitric oxide synthase and lipoxygenases |
| DNA synthesis, replication and repair | Ribonucleotide reductases, DNA polymerases, DNA glycolsylases, DNA primases, DNA helicasess and DNA endonucleases. Dihydropyrimidine dehydrogenas |
| Collagen synthesis | Proline hydroxylase |
| Electron transport and respiratory chain | Cytochrome C oxidase, Cytochrome b, cytochrome c1, Cytochrome oxidase P540, NADH dehydrogenase, aconitase, citrate synthase, Succinyl dehydogease, cytochrome reductase, Complex I-III, rieske protein, NADH ferrocyanide oxidoreductase |
| Adrenoxin | Steroid hydoxylation |
| Antioxidant defence | Catalase |
| Response to oxidative stress | Glutathione peroxidase 2, lactoperoxidase |
| Amino acid metabolism | Tryptophan pyrrolase, Phenaylalanine hydroxylase, deoxyhypusine hydroxylase |
| Carnitine biosynthesis | α-ketoglutarate (αKG)-dependent oxygenases |
| Synthesis of thyroid hormone | Thyroid peroxidase |
| Drug detoxification | Cytochrome P450 , NADPH cytochrome P450 reductase |
| Prostaglandin thromboxane synthesis, inflammation and response to oxidative stress | Cyclooxyenase |
| microRNA biogenesis | DiGeorge Syndrome Critical Region Gene 8 |
| Ribosome function and tRNA modification | ABCE1, CDKRAP1, TYW1 and CDKAL1, Methylthiotransferase |
| Haeme biosynthesis | Ferrochelatase |
| Apoptosis and oxygen transport in the brain | Neuroglobin |
| Purine metabolism and synthesis | Xanthine oxidase, amidophosphoribosyltransferase |
1.2. Definition of Iron Deficiency
2. Deleterious Biological Consequences of Iron Deficiency