1.1. Introduction
Additive manufacturing has been a major breakthrough in construction technologies and has been considered “the third industrial revolution” [
1]. Additive manufacturing, commonly known as 3D printing, allows for building parts one layer at a time from a 3D computer model, allowing for rapid design optimization and customization. Because of these interesting properties, medical applications have been quickly developed: 3D-printed prostheses, implants, anatomical models, etc. [
2,
3]. The ease of use and speed of prototyping has even allowed for quick responses to medical needs during the COVID-19 pandemic [
4,
5,
6].
The rapid development of this technology has required the development of new materials capable of being printed, in particular plastics, but also metals, ceramics, and elastomers. Traditionally, the materials used for 3D printing in medicine are made of inert and acellular materials, such as plastics [
7]. Among those materials, some are bio-compatible and can thus be used for implantation [
8]; other materials are degradable and are used as guides for soft tissue reconstruction, e.g., breast reconstruction after cancer surgery [
9]. Recently, a new field of research in 3D printing has emerged: 3D bioprinting. Three-dimensional bioprinting uses 3D-printing technology to print cells and a supportive matrix (called bioink) altogether, ultimately printing a living tissue [
10]. Bioinks have been defined by Groll et al. as “a formulation of cells suitable for processing by an automated biofabrication technology that may also contain biologically active components and biomaterials” that could be resumed as cell-containing materials [
11]. Three-dimensional bioprinting, while promising, raises a large number of concerns and challenges, in particular the development of biocompatible bioinks and their integration into the human body; however, it seems to be a proper tool for complex tissue in in vitro modeling [
12]. Although bioprinting is mainly used for tissue engineering, it seems to us that this technology also has its place in research teams wishing to model the complexity of the cancer microenvironment: its heterogeneity, its mechanical environment, its metabolism, and its neoangiogenesis, all of which can, of course, be used for drug-screening purposes.
In this review, we will try to propose an easy approach that allows the implementation of bioprinting in a research team and more particularly in the context of cancer modeling. We will first summarize the main technical features necessary for the implementation of bioprinting: the choice of the bioprinter, the choice of the bioink, and the polymerization method. Secondly, we will detail how bioprinting allows us to refine cancer research, notably by adding a cellular and mechanical complexity that 2D culture cannot provide. The aim is not to be exhaustive but rather to be comprehensive.
1.2. Bioprinting Technologies
The first bioprinting technique was described in 2003 by Boland et al., who used an inkjet-based technique to print 2D tissue constructs [
13]. Since this first experiment, numerous bioprinting technologies have been created and can be classified into three main categories depending on the type of cell deposition: drop-based (e.g., inkjet or laser bioprinting), filament-based (e.g., extrusion bioprinting), and plane-based (e.g., digital light processing (DLP)/stereolithography (SLA) bioprinting) (
Table 1).
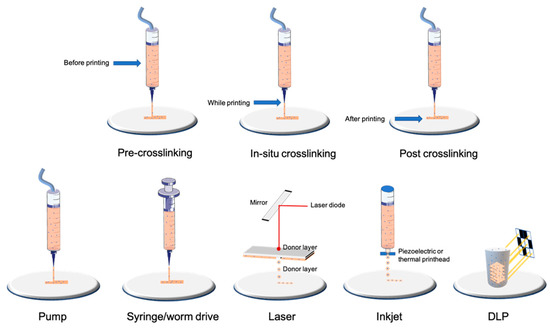
Nowadays, the most-used technology is the filament-based one, with different extrusion mechanisms: pneumatic, piston, and screw-driving (
Figure 1). Extrusion-based techniques resulting in filament deposition are nowadays the most used as they can quickly produce scaffolds of a resolution down to 100 μm in an affordable and relatively simple way [
14].
Figure 1. Examples of bioprinting and crosslinking technologies.
In our opinion, nowadays, this technology is the easiest to implement; many manufacturers offer machines with multiple extrusion printheads (some printheads may even use inkjet-based printing techniques (see below for details)) all in a tabletop format and with a user-friendly interface at reasonable prices. This technology is also compatible with almost all bioink formulations [
15].
Droplet-based techniques are consistent with the discontinuous printing of microdroplets and thus a high resolution (for review, see [
16]). Inkjet printing is the most common technology used for droplet generation and consists of a piezoelectric or thermal actuator that allows the precise deposition of the droplets down to 50 μm [
17,
18]. Laser-based droplet deposition allows single-cell deposition and, as a non-contact method, is responsible for low shear stress and thus excellent viability; the drawback is the expensive price of this type of 3D printer [
19,
20]. There are also other less-used approaches, such as acoustic- or valve-based droplet bioprinting technologies [
21,
22]. Even if the droplet generation (surface tension) and breaking combined with the force with which it will be projected onto the printing plate can reduce cell viability, drop-based approaches allow higher cell viability than filament-based ones (>85%).
This technology, although it brings a precision that extrusion-based ones cannot have, only allows the printing of 2D patterns. This can be useful to precisely include cells in a pre-existing 3D matrix but not for large-scale constructions.
Plane-based 3D printing is mainly consistent in DLP and SLA technology (for review, see [
23,
24]). In SLA technology, photopolymerization is achieved through a laser beam scanning the surface of a liquid bioink, whereas in the DLP technique, polymerization is achieved by a digital micromirror device (DMD) or by a liquid crystal display (LCD) [
25]. Volumetric bioprinting is a technique derived from those light-based techniques and can enable the creation of entire objects at once, which allows free-form architecture bioprinting that cannot be achieved with other technologies [
26].
Those techniques have a high resolution down to 25 μm and speed in producing large and complex volumes; however, this technique requires a large volume of bioink, a significant part of which will not be polymerized [
27].
Despite these interesting characteristics, particularly the speed of printing large volumes and the precision, it is not the easiest to implement this technology, particularly because of the lack of compatible bioinks and its running cost. It is, however, interesting for printing complex microfluidic structures.
2. Characterization of Cells after Bioprinting
To evaluate the success of a bioprinting model, one of the most important parameters to assess is the viability and metabolic activity of the cells. Indeed, it is necessary to find the adequate printing parameters that allow for obtaining the structural integrity of the hydrogel so that it is reproducible and especially viable. These parameters must be determined for each type of bioink and even for each concentration. Printing parameters, such as the bed or cartridge temperature, pressure, and printing speed, will modify the viscosity of the gel, which will affect the shear stress exerted on the cells and, therefore, their viability. This is also impacted by the way the hydrogels are crosslinked.
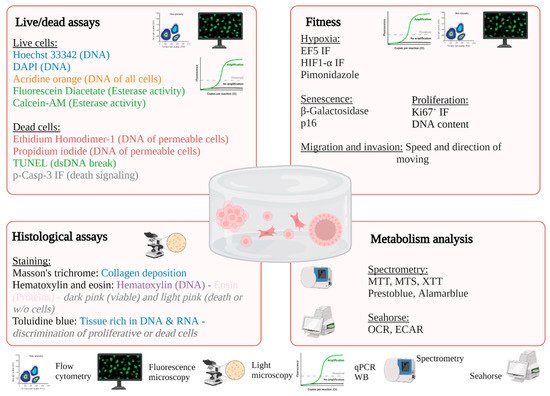
A plethora of techniques is available to characterize cells after bioprinting to determine the size and organization of the constructs, cell viability, and metabolism and the level of gene and protein expression [
98,
99] (
Table 3,
Figure 3). The size and shape of the constructs must be adapted to the technique used. For example, microscopic analysis does not require many cells, in contrast to cytometry, molecular biology technics, or spectrometric analysis. After adaptation, the usual techniques used in conventional 2D culture can be applied.
Figure 3. Examples of possible analysis of 3D bioprinted constructs.
2.1. In Situ Characterization of Cells
The advantage of using techniques where the cells are embedded in the hydrogel allows for avoiding artifacts related to the dissociation of the hydrogel.
2.1.1. Light Microscopy
Microscopy is particularly interesting in the characterization of hydrogels because it allows the structure of the construct to be preserved, as well as the cell–cell interactions. It allows access to the size and morphology of cells that could assemble into spheroids or in a native tissue organization. Phase-contrast microscopy allows for monitoring cell proliferation and growth over time without inducing toxicity [
100,
101,
102]. However, because the cells are alive, the acquisition time should not be too long to avoid inducing cell death. This technique is only possible for optically transparent hydrogels. For example, the cell-ink bioink composed of alginate and cellulose nanofibril is opaque and does not track cells without prior fluorescent labeling or end-point histological analysis.
Histological analysis requires sample preparation, including fixing, cutting, and staining [
101,
102,
103]. The preparation steps for sectioning are very important. Dehydration for paraffin embedding tends to shrink the size of the sample and is therefore not be recommended for structural or organizational measurements [
104]. In addition, if the hydrogel pores are not completely filled with paraffin, this will promote folding during sectioning and detachment of the sample from the section. However, the advantage of this technique is the possibility of having thin sections (up to 5 µm thick). In contrast, cryosection preserves the hydrogel structure, particularly with polyvinyl alcohol (PVA) and optimum cutting temperature (OCT) preparation. However, the sections are thicker, and more aspecific markings can be observed with a protein-based cryoprotectant solution [
105]. Using resins favors the preservation of structures but makes it more difficult to perform histological stains [
106]. Finally, it is possible to proceed directly to histological staining without cutting to visualize the cells on the surface of the hydrogel. Depending on the structures of interest, different stainings are available: Masson’s trichrome (TM) stains collagenous structures in blue (fibrosis, for example); hematoxylin (DNA) and eosin (proteins) illuminate viable zones in dark pink and dead zones in clear pink; and, finally, toluidine blue highlights the zones rich in RNA and DNA. Trypan blue is used to stain dead cells [
107]. Quantification of chromatic staining can be difficult on thick samples, so the use of fluorescence microscopy is a good alternative.
2.1.2. Fluorescence Microscopy
Fluorescence microscopy is used to label subcellular structures, such as the cytoskeleton (F-actin), mitochondria (MitoTracker), nuclei (Hoechst), or other types of organelles or proteins [
108,
109,
110,
111]. Standard immunofluorescence or biomarker labeling protocols can be applied to the hydrogel, although the times of the different labeling steps should be increased or even improved using mechanical agitation or a vacuum. Observation of the organization and viability of cells as a function of the position or shape of the hydrogel is only possible under microscopy. Using markers or antibodies coupled to fluorescent probes, it is possible to determine whether cells are dying (p-casp3), proliferating (KI67
+ or DNA), entering in senescence (p16 or β-galactosidase), or in a hypoxic environment (HIF1-α, EF5, pimonidazole). Numerous fluorescence assays for dead/live cells are described in
Table 2; however, the most commonly used combination of fluorochromes is calcein AM stain for esterase activity (live cells) and propidium iodide for permeable and therefore dead cells. It is possible to combine one of these two markers with Hoechst3342 or DAPI; however, this is not possible in all types of hydrogels, such as alginate, which shows strong auto-fluorescence from the UV channel. An easy-to-use marker for studying cell morphology is phalloidin labeling of F-actin, which is particularly interesting in models for studying mechanotransduction as a function of support stiffness, for example [
107,
112,
113,
114].
For high-resolution microscopy, confocal imaging is the reference method for studying cells embedded in the hydrogel. The disadvantage is the necessity to print thin film constructs on suitable substrates. Indeed, without a clearing technique, only 100 µm-thick constructs can be imaged. Furthermore, these hydrogels should preferably be printed on glass coverslips to favor high-resolution imaging. The risk is that the hydrogel may become detached; to mitigate this, the silanization of the coverslips allows the covalent bonding of the gel with its support. To limit the constraints of confocal imaging, other microscopy techniques have been developed, such as light sheet imaging. It is thus possible to image large objects without a physical section with limited phototoxicity [
108,
109,
110,
111].
2.1.3. Electronic Microscopy
Electron microscopy provides nanoscale imaging, either scanning for the sample’s surface or transmission for the internal structures [
102,
115]. These techniques allow the study of cell–cell or cell–ECM interactions but also cell death. However, the sample preparation steps can change the structure of the sample.
2.2. Characterization of Cells after Isolation or Lysis
2.2.1. Molecular Biology
For many applications in tissue engineering, it is necessary to be able to extract DNA, mRNA, or protein in order to monitor different cellular parameters such, as differentiation or certain functions. It is also possible to determine proliferation and viability via the measurement of DNA concentration. This technique is interesting since it allows for knowing the number of cells per gel but also for normalizing the data obtained to the number of cells. However, it is critical to find the right technique to lyse the cells to recover the full amount of DNA. For example, for alginate gels, it is possible to use a commercial solution, the purelink genomic DNA mini kit [
133]. For GelMA or agarose, the use of EDTA associated with proteases allows the recovery of cells from the gel in order to assay the DNA [
113,
114,
134,
135,
136,
137].
Conventional methods for 2D cell culture rely on two methods: either via the use of phenol/chloroform or with commercial kits using silica membranes in spin columns [
138]. However, the inclusion of cells in hydrogels makes this step more difficult, and it presents more challenges that are technical. Indeed, the classical RNA extractions often do not allow for obtaining RNAs in sufficient quantity and/or quality for the subsequent performance of RTqPCR. Köster’s team conducted a study to investigate homogenization methods and RNA extraction techniques based on the most commonly used hydrogels (alginate, gelatin, and agarose) on hMSC cells [
139]. For this purpose, four homogenization techniques are deployed. Regardless of the type of hydrogel, homogenization techniques using liquid nitrogen or a rotor stator should be excluded, as the yield of RNA is very low. In contrast, the amount of RNA is much higher for techniques using the micro-homogenizer or enzymatic/chemical digestion. The technique of frozen liquid nitrogen crushed by an electric crusher seems to be relevant for GelMA-type homogenization [
113,
134]. For extraction, Köster’s team shows that conventional commercial kits using silica membranes in spin columns do not provide a correct RNA yield for hMSC in alginate, gelatin, and agarose hydrogels [
139]. However, other teams obtain satisfactory results with agarose-based or alginate hydrogels [
114,
140]. Hot phenol (HP), TRIzol (TR), cetyltrimethylammonium bromide (CTAB), and LiCl (LC) techniques have a better RNA yield, but the LiCL technique gives poor PCR results (e.g., dominating additional band, PCR product with incorrect size or no PCR product). For the same reasons, the TRIzol technique is not adapted for alginate gels for Köster’s team but it is for Ewa-Choy’s or Sbrana’s Teams [
118,
133]. Hot phenol and CTAB seem to be the most suitable techniques; hot phenol gives the best RNA yield, and CTAB gives the best RNA quality (equivalent to 2D culture) and low endpoint Ct values ~20.
2.2.2. Flow Cytometry
It can also be interesting to isolate cells to either promote cell expansion or analysis using flow cytometry, as this allows many cells to be analyzed very quickly. For this purpose, enzymatic degradation is possible for matrices derived from natural products, such as collagenase for GelMA or collagen hydrogels, hyaluronidase for hyaluronic acid-based gels, or alginate lyase for alginate hydrogels. Some materials can also be degraded by physical techniques, such as photo-degradation [
141]. This step is critical because a too-prolonged enzyme treatment can induce significant cell death or even alter the membrane receptors. The limitation of this technique also lies in the fact that a large hydrogel is required to recover the necessary number of cells after degradation [
142]. Then, standard labeling protocols such as 2D culture can be used. Flow cytometry allows quantitative measurements of many parameters simultaneously, such as viability, proliferation, cell cycle, and uptake of anti-cancer agents. As for microscopy, live/dead tests based on calcein AM and ethidium are the most commonly used, with propidium iodide or BrdU for the cell cycle. It is also interesting to use this technique to identify subpopulations or maintenance of a phenotype, such as chronic lymphocytic leukemia cells on CD5
+CD19
+IgM
+ markers [
118]. The disadvantage of flow cytometry compared to microscopy is the loss of spatial information. To compensate for this, Beaumont’s team developed a protocol based on the diffusion gradient of Hoechst 33342, which makes it possible to discriminate between internal and peripheral cells according to the intensity of Hoechst [
143].
Table 3. Characterization technology of bioprinted constructs. Value of 3D bioprinting for cancer modelling. + for pros and − for cons.
| Methods |
Description |
Pros and Cons |
Markers |
REF |
| Microscopy |
| Light |
Phase contrast |
Monitoring of proliferation and morphology of cells |
+: • Nondestructive
• No markers are added
• Low cost
• Easy with transparent gels (GelMA, matrigel)
−: • No possibility to identify subcellular structures
• Difficult with opaque or non-transparent gels (e.g.,: alginate with nanocellulose) |
Not suitable |
[100,101,102] |
| Bright field |
The transmission of light is more or less attenuated depending on the density or marking of the sample |
+: • Suitable for large samples
−: • Requires histological staining
• Preparation of sample
• Quantification of thick sample |
Hematoxylin–eosin
Masson’s trichrome
Trypan blue |
[101,102,103] |
| Fluorescence |
LSM
Epifluorescence
Confocal |
The use of a fluorescent marker is necessary to highlight a subcellular structure; possibility of monitoring structures over time (if vital markers) |
+: • Monitoring of many possible structures
−: • Requires cutting for oversized constructions for epi and confocal microscopy
• Need to fix for certain markers
• Important autofluorescence for chitosan or alginate/cellulose hydrogels in UV |
Live/dead staining
Or calcein AM/propidium iodide
Or ethidium homodimer
Active-caspase3/7 green
Hoechst 33342
HIF1-α, Ki67 |
[108,109,110,111,144] |
| Electronic |
Scanning |
Surface is scanned with a beam of electrons, emitted signal provides images |
+: • High resolution
−: • The preparation procedure is tedious
• Frequent preparation artifacts (collapse) |
Not suitable |
[102] |
| Transmission |
The part of beam of electrons is transmitted into specimens allowed to obtain images |
Not suitable |
[102,115] |
| Flow cytometry |
| Flow cytometry |
Analysis of physical parameters (size and granularity) for each cell but also the level of fluorescence |
+: • Quantitative analysis
−: • Disaggregation can be a problem
• Necessity to have a large cell number due to loss of cells during dissociation |
7-AAD
CFSE |
[102,139] |
| Spectroscopy |
| Spectrometry or fluorimetry |
Production or utilization of a fluorescent or chromatic compound |
+: • Well-described for 2D culture and frequently used
• Can be used for kinetic monitoring
−: • Ensure that the efficiency is adapted for 3D |
ACP, LDH, prestoblue, alamar blue, DNA content |
[112,119,120,121] |
| Molecular biology |
RTqPCR
Western blot |
Quantification of gene expression at mRNA or protein level |
+: • Quantitative analysis
• Easier by using the enzymatic method on natural inks (e.g., collagenase for GelMA or ColMA, hyaluronidase for hyaluronic acid)
−: •Adaptation of the homogenization and extraction protocol to obtain an adequate quantity and quality of RNA/proteins for analyses |
Bax/Bcl2
HIF1-α, Ki67 |
[103,115,118] |
| Metabolism |
| GC–MS (Gas chromatography–mass spectrometry) |
Detection of molecules of interest according to their mass/charge ratio after ionization |
+: • Considerably less cellular material compared to NMR, high sensitivity,
−: • Use of radioisotopes, complex sample preparation, high cost |
13C-Glucose |
[129,132] |
| NMR (nuclear magnetic resonance) spectroscopy |
Determination of the composition of a sample by applying a magnetic field via the orientation of the nuclear spins of the atoms |
+: • High reproducibility, sample can be analyzed directly, low cost
−: • Use of radioisotopes, low sensitivity |
[130,131] |
| PET scan (positron emission tomography) |
Injection of a radiographic tracer and monitoring by imaging to detect localization of [18F]FDG |
+: • Classically used in medicine, monitoring over time
−: • Low resolution (1.5 mm) |
[18F]FDG |
[120,125] |
| Seahorse |
Quantification of the oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR) |
+: • High sensitivity (from 5000 cells, theoretically), possibility to test many conditions in parallel
−: • Difficulties in normalizing results, limited number of injections, limited sample thickness |
Not suitable |
[126,128] |