Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Karine Elena Rodriguez Fernandez and Version 2 by Amina Yu.
Psoriasis is a chronic autoimmune and inflammatory skin disease associated with physical and psychological burdens characterized by erythematic plaques with adherent shiny scales. The country-specific prevalence of psoriasis varies from 0.14% (95% uncertainty interval 0.05% to 0.40%) in east Asia to 1.99% (0.64% to 6.60%) in Australasia. Additionally, the prevalence is high in western Europe (1.92%, 1.07% to 3.46%), central Europe (1.83%, 0.62% to 5.32%), and North America (1.50%, 0.63% to 3.60%).
- psoriasis
- monoclonal antibodies
- therapeutic drug monitoring
- pharmacokinetics/pharmacodynamic models
1. Introduction
Psoriasis is a chronic autoimmune and inflammatory skin disease associated with physical and psychological burdens characterized by erythematic plaques with adherent shiny scales [1]. The country-specific prevalence of psoriasis varies from 0.14% (95% uncertainty interval 0.05% to 0.40%) in east Asia to 1.99% (0.64% to 6.60%) in Australasia. Additionally, the prevalence is high in western Europe (1.92%, 1.07% to 3.46%), central Europe (1.83%, 0.62% to 5.32%), and North America (1.50%, 0.63% to 3.60%). Its age of onset shows a bimodal distribution, with peaks at 30–39 years and 60–69 years in men, and 10 years earlier in women [2]. The phenotypes of this disease are plaque psoriasis or psoriasis vulgaris, guttate psoriasis, inverse psoriasis, and erythrodermic psoriasis, which differ in terms of their clinical and morphological characteristics [3][4][5][3,4,5]. In addition, nail psoriasis is reported to affect more than half of the patients [6].
1.1. Pathophysiology of Psoriasis
A complex and not fully understood pathogenesis is exhibited in psoriasis. External factors can trigger an interaction between skin cells, pro-inflammatory immunocytes (i.e., tumor necrosis factor (TNF)-α and interferon (IFN)-α), and biologic signaling molecules in genetically predisposed individuals [7][8][7,8]. This interaction stimulates the myeloid dendritic cells (mDC) in the lymph nodes to release interleukin (IL)-12 and IL-23 to promote the cellular immune response of T helper lymphocytes (Th) type 1 (Th1), 17 (Th17), and 22 (Th22) T cells. Activated Th migrate to the skin guided by a gradient of chemokine and produce abundant psoriatic cytokines (for i.example., IL-17, IFN-γ, TNF-α, and IL-22). The cytokine-mediated effects on keratinocytes influence typical psoriatic inflammation [9][10][11][12][13][9,10,11,12,13]. Molecular and genetic onstudies in specific psoriasis phenotypes have identified different inflammatory pathways that may coexist and evolve over time. The identification of the main inflammatory pathways through individual molecular descriptors represents a future step to guide personalized therapy [14]. In this sense, different classes of possible biomarkers have been explored in psoriasis (Figure 1), but further replication and validation are required [15][16][17][15,16,17].
1.2. Clinical Endpoints of Psoriasis
The severity of psoriasis will be determined by the extent of the disease, the location of the lesions, the degree of inflammation, and the impact on quality of life. According to the most important clinical guidelines (Figure 1), the evaluation of psoriasis severity and the levels of its treatment responses is generally based on the percentage of the total Body Surface Area (BSA) affected, Psoriasis Area Severity Index (PASI), Physician Global Assessment (PGA), and Dermatologic Life Quality Index (DLQI) [18][19][18,19].
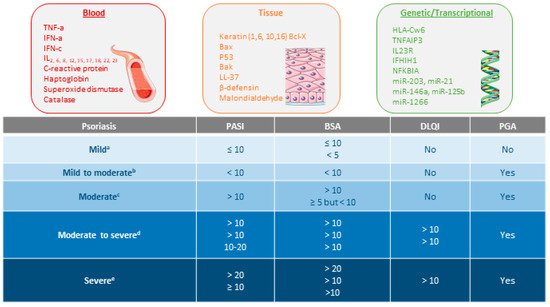
Figure 1. Types of biomarkers in psoriasis and psoriasis severity criteria according to several consensus guidelines or clinical associations. References supporting the consensus for a [20,21], b [22], c [21,22], d [20,22,23], and e [21,22,24].
2. Pharmacokinetic/Pharmacodynamic Properties of Monoclonal Antibodies in Psoriasis
Despite the increasing number of therapeutic monoclonal antibodies (mAb) on the market and in the drug development process for psoriasis treatment, the pharmacokinetic (PK) and pharmacodynamic (PD) properties of these molecules are more specific. In this regard, non-linear mixed-effects modeling allows for the accurate quantification of the central tendency and the different sources of the variability of mAb by considering data from all individuals simultaneously. The aims of this review are (i) to describe the main factors involved in the management of psoriasis disease with biological therapy, and (ii) to provide insights into the role of therapeutic drug monitoring (TDM) through population PK and PK/PD modeling strategies in the mAb treatment of patients with psoriasis.
2.1. Pharmacokinetic Properties
Monoclonal antibodies are heterodimeric glycoprotein macromolecules of type-G immunoglobulin recognizing a single epitope on a target antigen in a bivalent manner [20][25]. They are produced and engineered by hybridoma technology, developed for the first time by Köhler and Milstein in 1975 [21][26]. Due to their molecular size and their three-dimensional conformation, the PK and PD properties of mAbs are considerably different compared with those related to small-molecule drugs (SDM) [22][27].
The low permeability and high degradation of mAbs throughout the gastrointestinal tract lead to intravenous, subcutaneous, or intramuscular administration [23][28], and no significant improvement has been published to overcome the limitations of the oral administration of mAbs. Consequently, the most frequent routes of the administration of mAb in psoriasis follow intravenous (IV) or subcutaneous (SC) injections [24][29]. Regarding the distribution and tissue infiltration, mAbs can easily move from the SC space most probably via diffusion and/or convection through lymphatic capillaries, and they can be able to reach the intracellular space of targets beyond systemic circulation by pinocytosis or by receptor-mediated endocytosis [25][30].
The large size and physicochemical properties (charge and hydrophobicity) explain the distribution of mAbs mainly in the vascular and interstitial fluids. Usually, tissue distribution represents 5 to 15% of the total amount of mAb, and distribution into the brain is quite restricted (0.1%) [26][31]. A significant fraction of mAb in the body may be found if mAb-tissue target binding occurs with high affinity. Therefore, large apparent volumes of distribution in steady state (Vss) could be estimated for mAbs. In cases where the binding capacity of tissue is limited, nonlinear distribution is more probable and Vss decreases in a dose or concentration-dependent manner [27][32].
In regard to mAbs metabolism and excretion, the impacts of the renal and biliary pathways are insignificant [28][33]. Due to the null role of enzymatic processes related to the metabolism and excretion of mAb, the interaction with other substrates of these enzymes is negligible [29][30][31][32][34,35,36,37]. mAbs exhibit specific and non-specific types of binding, depending on the fragment of the antibody. The first one occurs when the antigen-binding fragment (Fab) attaches to the target antigen. The second one appears after the fragment crystallizable (Fc) region binds to cell surface receptors, such as the Fcγ receptor (FcγR) on the immune effector cells and the neonatal Fc receptor (FcRn) on different cell types, as well as components of the complement system (for i.example., complement C1q) [20][25]. For such reasons, mAb distribution can be directly influenced by the density and expression of the target antigen. The two parallel metabolic pathways, for i.example., specific and non-specific. are involved in mAb disposition, and their impact changes over time based on the available free mAb in the plasma and the dose administered. Metabolism through the reticuloendothelial system via pinocytosis/proteolysis represents the linear and non-specific clearance, which may be relevant at certain dose levels due to the larger endothelial surface area in the gut, muscle, and skin [33][38]. The specific pathway is initiated after the internalization of the receptor–drug complex, which allows the drug to enter the cell and then be inactivated by cytoplasmic endosomes. However, FcRn can bind IgG and mAbs at the acidic pH conditions of the lysosome, escape from proteolysis, and be directed back to the cell membrane [34][35][36][39,40,41]. Both pathways have been mechanistically described in population PK models through a target-mediated drug disposition (TMDD) approach and its quasi-equilibrium or rapid binding approximations, quasi-steady-state approximation, and even simpler Michaelis–Menten kinetics.
One more key aspect in the PK of mAb is the rescue from lysosomal degradation by binding to FcRn in endothelial cells, which is crucial for the long half-life and low clearance rate reported for most therapeutic mAbs [37][38][42,43]. These mechanisms result in clearance values ones to f mAbs for psoriasis that range from 90 to 560 mL/day, leading to half-lives between 11 and 30 days. Several covariates have been identified in PK studies to partially explain inter-individual differences in mAb exposure, such as FcRn and FcγR gene expression, genetic polymorphism, target properties, and covariates associated with increased clearance, such as the generation of antidrug antibodies (ADA), low serum albumin and high serum C-reactive protein levels (CRP), gender, and high body weight (BW) [22][25][27,30].
2.2. Pharmacodynamic Properties
The mAbs for treating psoriasis are designed to block either the specific receptors or soluble mediators of the main pathways in the progress and chronicity of psoriasis, including TNF-α, IL-12/23, and IL-17 [11] (Figure 12). The PD effect of mAb is delayed to the time course of its plasma concentrations, which has been described using PK/PD models, such as indirect responses and transduction models, in order to describe the exposure–response (E–R) relationship [39][40][44,45]. The following parameters are mostly determined: kin, formation rate of psoriatic skin lesions; kout, remission rate of psoriatic skin lesions; Emax, maximum mAb effect; and EC50 or IC50, serum mAb concentration causing 50% of the maximum effect.
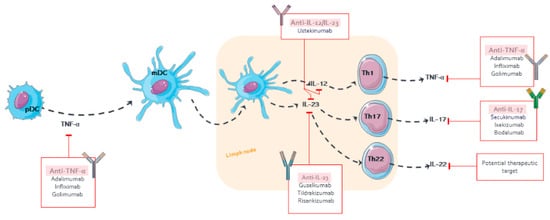
Figure 12.
Pharmacological targets of monoclonal antibodies in psoriasis.
Compared with SDM, mAbs offer therapeutic exclusivity, a higher safety profile, and an increase in clinical efficacy [41][42][46,47]. The relative change in the PASI of patients receiving novel mAbs (guselkumab and brodalumab) has reached 90–100% PASI reduction, which has led to an adjustment of the primary endpoint of PASI75 to PASI90 or PASI100 in clinical trials [43][48].
3. Monoclonal Antibody Approved for Psoriasis
The selection of the optimal treatment for psoriasis depends on the severity of the disease [44][67]. Mild or limited-extent psoriasis is managed by topical treatment, while the moderate to severe types usually require a combination of phototherapy and systemic therapies [45][68]. Biological agents, such as mAb, have been the most successful approach in the management of this disease in the last decade.
The use of mAbs is indicated in psoriasis when (i) effective control of psoriasis is not achieved with oral and phototherapy treatments, (ii) in patients who have rapid regrowth (3 months or less) after suspending any treatment, (iii) when higher doses of conventional systemic drugs are required with the increased associated risk of adverse effects, (iv) in patients with comorbidities for which the use of systemic agents, such as methotrexate or cyclosporine, are contraindicated, (v) when a patient is unable to tolerate the traditional systemic therapy, or (vi) the patient is at high risk of toxicity with methotrexate, cyclosporine, acitretin, or phototherapy, even in the absence of analytical alterations [46][23].