Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Frederique VEGRAN and Version 2 by Rita Xu.
Splicing is a phenomenon enabling the excision of introns from pre-mRNA to give rise to mature mRNA. All the 20,000 genes of the human genome are concerned by this mechanism. Nevertheless, it is estimated that the proteome is composed of more than 100,000 proteins. How to go from 20,000 genes to more than 100,000 proteins? Alternative splicing (AS) is in charge of this diversity of proteins. AS which is found in most of the cells of an organism, participates in normal cells and in particular in immune cells, in the regulation of cellular behavior. In cancer, AS is highly dysregulated and involved in almost all of the hallmarks that characterize tumor cells.
- alternative splicing
- cancer cells
- immune cells
1. Introduction
It has long been estimated that the human genome was composed of nearly 300,000 genes. With the advent of new sequencing technologies, it is now assumed that reswearchers hold around 20,000 genes. This state of affairs may seem surprising at first. Indeed, many species such as zebrafish or the nematode worm hold as many genes or more. Thus, there would be a paradox that the number of genes is not representative of the biological complexity of an organism. Several mechanisms that can address this problem have been discovered, including alternative splicing (AS) of pre-messenger RNAs. This phenomenon makes it possible to obtain from a single gene several functional proteins. If at the time of the discovery of AS it was estimated that about 10 to 15% of human genes produced multiple transcripts, it is now established by RNA sequencing that it is actually 95 to 98% of genes that generate multiple isoforms. AS which puts an end to the “a gene, a protein” paradigm leads to a reconsideration of the very conception of what a gene is.
If AS grants healthy cells flexibility and adaptability according to a given environment or stimulus, it can sometimes be deleterious and contribute to their transformation into cancer cells. Indeed, more and more studies show that AS has a preponderant role in cancer development. On the other hand, it has been shown that AS is involved in the homeostasis and differentiation of immune cells and especially those of acquired immunity. Through the different aggression of non-self to which they must respond, these cells are brought to evolve and adapt in as many different environments as a ganglion, blood, or lymphatic circulation or even a tumor can be.
2. Alternative Splicing and Cancer Immunotherapy
Immunotherapies are therapeutic strategies very effective in around 15–20% of patients. The presentation of tumor neoantigens seems to be one of the predictive markers of immunotherapies response (Figure 1).
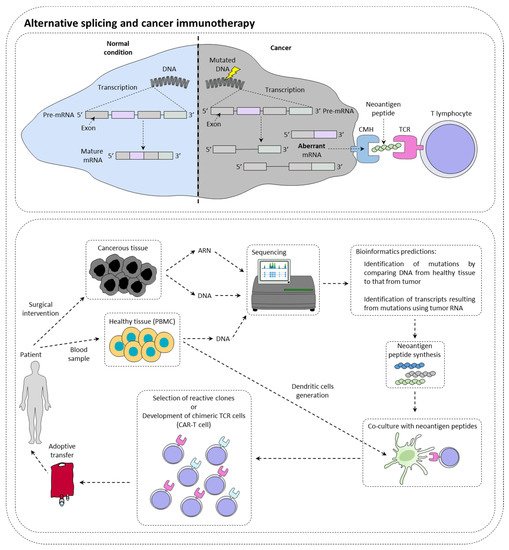
Figure 1. Alternative splicing and cancer immunotherapy. Alternative splicing could have a direct impact on immunotherapy response by creating neoantigen peptide that could be recognized by immune cells. This is currently under investigation for the production of CAR-T cells.
2.1. Alternative Splicing Can Increase Immunotherapy Response
Immunotherapy targeting immune checkpoints such as PD-1 (programmed cell death 1) or CTLA-4 (cytotoxic T lymphocyte-associated protein 4) is a real revolution in the treatment of many cancers [1]. In this context, it has been observed that tumors with a high mutational load have more tumor peptides at their disposal surface [2]. However, while many studies show a positive correlation between the level of tumor mutation and the response to checkpoint inhibitors [3], others contradict them by showing that sometimes highly mutated patients do not respond to treatment, while other patients with few mutations respond to treatment [4]. In this context, it has been shown that patients with somatic mutations generating new splice sites respond better to immunotherapies targeting PD-L1 [5]. This type of mutation could then be a new clinical biomarker of choice in the response to checkpoint inhibitors. New technologies such as RNA sequencing or mass spectrometry (MS) now make it possible to identify tumor neo-antigens.
With the advent of cell therapy techniques, it is now possible to target these antigens by selecting a cytotoxic lymphocyte clone specific for a given tumor antigen, then amplifying it and performing an adoptive transfer of these cells. It is also conceivable to use cytotoxic lymphocytes that have been reprogrammed genetically to express a chimeric TCR that makes it possible to recognize a specific tumor antigen (CAR-T cells (chimeric antigen receptor T cells). Nevertheless, there are still many obstacles to the implementation of therapeutic strategies to specifically target these antigens. It is first of all crucial to determine whether the tumor neoantigen is specific to the tumor.
2.2. Alternative Splicing Generates Neoantigens
It has recently been demonstrated in a study of more than 8705 patients, that tumors had more than 30% more AS events than healthy tissue samples [6]. Focusing on ovarian and breast tumors that also had MS data and transcriptomic data, 68% of tumors were found to have one or more tumor neo-epitopes derived from the tumor. In contrast, only 30% of tumors have tumor neo-epitopes from somatic mutations. Thus, it highlights the importance of analyzing AS events within tumors to identify new targets. Moreover, another study using MS demonstrated that most tumor antigens from two murine lines and seven primary human tumors originated from the translation of coding exons out of the reading frame, but also from non-coding regions of the DNA. All of these events can most likely originate from the deregulation of AS in these tumors [7].
Since AS is tissue-specific regulated, it is essential to analyze whether the AS event found within the tumor does not occur elsewhere in the body and not just in the healthy tissue surrounding the tumor [8]. The transcriptomic databases available for each organ could answer this problem in order to avoid any nonspecific immune responses that could have dramatic effects. In addition, it is still difficult to determine AS events at the subclonal level in tumors. Single-cell RNA sequencing may be the key to overcoming this technical difficulty, but the starting material used for this technique is currently too weak to expect anything other than the expression of highly represented transcripts in cells [9]. However, new algorithms appear promising in identifying alternative transcripts from relatively low coverage RNA sequencing [10]. Regarding MS, other problems come to oppose the use of tumor neo-antigens from AS. Indeed, the detection threshold is often very high and requires a large quantity of cells to detect minor events of AS at the protein level [11]. In addition, it is important to test the immunogenicity of tumor neo-epitopes from AS. Indeed, in melanoma and glioblastoma, 51.7–66% of neo-epitopes presented by class II MHC generate a CD4 + T cell response, and only 16–43% of those presented by class I induce a response from the cytotoxic lymphocytes. Factors such as the specificity of peptides for MHC, their abundance but also the effectiveness of their presentation should be taken into account. Nevertheless, it appears that many neo-epitopes derived from AS have an immunogenic capacity [12][13][14][12,13,14]. Huge progress has been made in cancer immunotherapies over the past decade [15]. Neo-epitopes from tumor-related AS are slowly being considered to develop promising new cell therapies for patients.
2.3. Alternative Splicing Can Limit Inhibitor of Checkpoint Inhibitor Response
AS may also limit the effects of immune checkpoint inhibitor-based immunotherapies. Recently, a PD-L1 splice variant has been identified in several cancers. It appears to be secreted and accumulates in the tumor microenvironment [16]. This variant has been shown to be involved in anti-PD-L1 resistance. CTLA-4 is another checkpoint inhibitor that has two isoforms: a membrane mCTLA-4 and a secreted form sCTLA-4. sCTLA-4 is a splicing variant devoid of exon 3 [17]. Studies have shown that both variants are expressed in tumor cells and are associated with immune escape from tumors [18][19][18,19]. Both bind to CD80 and CD86 on antigen-presenting cells (APCs) inducing a feedback of T cell activation [20]. At present, both variants have shown immunosuppressive activity but their prognostic role remains to be investigated.
2.4. Alternative Splicing May Limit the Efficiency of CAR-T Cells
The CD19 antigen is expressed on the surface of the majority of B cells in acute lymphoblastic leukemia. Cell therapy using chimeric TCR T cells specifically recognizing this antigen gives very good results. However, in 10 to 20% of cases, a relapse is noted after this treatment due to a decrease in the expression of this epitope [21]. It has recently been discovered that most of these relapses were due to a CD19 AS [22]. Since the alternative isoform CD19Δex2 is not recognized by the CAR-T-19, this could explain the relapses observed in some patients. The modification of the AS of this transcript is under the influence of the splicing factor SRSF3, but the activation of the latter during the relapse remains misunderstood. However, this discovery has isolated a new target from AS that will prevent many relapses in people with this disease and treated with CAR-T cells.
3. Alternative Splicing in Immunity Modulation
In the immune system, alternative isoforms have been identified in physiological contexts and others in pathological contexts, both in the innate and acquired immune systems [23] (Figure 2). For example, in macrophages, 81 AS events were observed during LPS stimulation [24]. Dendritic cells also develop different AS patterns, depending on the status of their differentiation and maturity. Single-cell RNA-sequencing has shown that different AS patterns exist depending on the maturity and differentiation of dendritic cells exposed to LPS [25]. However, the functional impact of these different splicing profiles has not been elucidated [25]. For T lymphocytes, as many as 1319 AS events occur during TCR stimulation and 1575 during TCR commitment and CD28 costimulation [26], including genes involved in the immune response [27]. Finally, more than 90% of B-cell multi-exon genes undergo AS [28]. Nevertheless, the role of alternative isoforms in immunity remains unclear. Moreover, the role of AS has not yet been characterized in all cell types and even less for tumor-infiltrating cells. The complexity of this phenomenon can be highlighted by the few examples that rwesearchers present below.
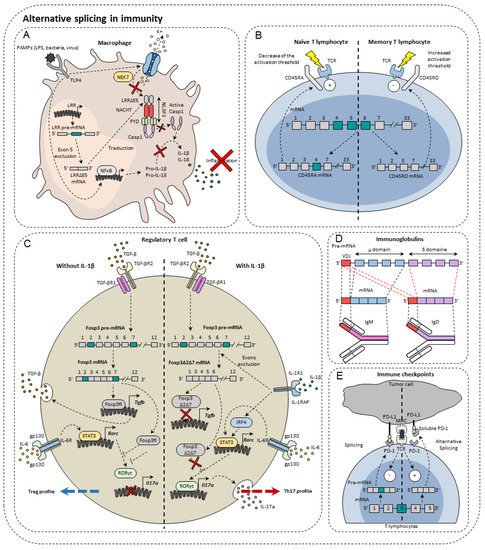
Figure 2. Alternative splicing in immunity. Alternative splicing has a direct impact on normal immune cells such as macrophages (A), T lymphocytes (B), regulatory T cells (C), immunoglobulin diversity (D), and immune checkpoints (E).
3.1. Immunoglobulins and B Cells
B cells are important players in adaptive immunity, by their propensity to secrete significant amounts of antibodies directed against a multitude of pathogens. In order to recognize many antigens, B cells rearrange the variable portion of their heavy and light chains within the bone marrow. These immature cells express on their surface IgM (immunoglobulin M). Once tested for their ability to respond to the self, conforming cells migrate to secondary lymphoid organs such as the spleen or lymph nodes. It is then the IgD that is expressed on the membrane surface. The reason for this transition from IgM to IgD has not yet been fully elucidated. However, it was observed that AS was responsible for the transition from the IgM isotype to the IgD isotype. The pre-mRNA encoding the IgM heavy chain contains the variable domain of recognition of VDJ antigens, the μ domain, but also the sequence encoding IgDs (δ domain). Upon the departure of B lymphocytes to secondary lymphoid organs, the AS that formerly led to the expression of IgM is directed towards a concomitant production of the two isotypes [29]. Although the exact role of this change is not yet fully known, it is known that the protein binding to the Zfp218 (zinc finger protein 318) RNA could be one of the players in the regulation of this AS. In fact, in follicular B cells, whose response depends on T lymphocytes, Zfp318 increases the amount of IgD at the expense of IgM [30]. Depending on their state of maturity, B cells are also able to secrete IgG, IgA, and IgE. It has been noted that IgE has several isoforms that appear according to different stimuli and the level of maturity of B cells [31]. AS is important in establishing the immune response by regulating its activation but also by orienting its differentiation.
3.2. Immune Cell Receptors
The importance of AS in macrophages has been demonstrated through the inhibition of the SF3A1 splicing factor that regulates the splicing of genes involved in the signaling pathway under TLRs [24]. Indeed, this leads to a decrease in the production of important regulators of this pathway, such as CD14, IRAK1 (interleukin-1 receptor-associated kinase 1), or IKKβ (inhibitor of nuclear factor kappa-B kinase subunit beta). This shows the importance of AS in the propensity of the innate system to activate in contact with the non-self through TLRs. It has also been demonstrated in macrophages and dendritic cells that there is a soluble form of TLR4 with a 144 base pair insertion between exon 2 and 3 leading to the appearance of a premature STOP codon. The appearance of this alternative isoform of TLR4 occurs during LPS stimulation. This results in decreased secretion of TNF-α and decreased activation of NFκB [32].
MyD88 (myeloid differentiation primary response 88) is a protein of great importance in the signaling pathway under TLRs. There is a long isoform of MyD88, MyD88L, that is responsible for the activation of innate immunity and a shorter isoform MyD88S, which instead restricts the immune response by blocking the signaling pathway. It was observed in this context that SF3A1, SF3A2, SF3A3, and SF3B1 splice factors were essential for the formation of the long form of MyD88 and that their genetic invalidation caused an inhibition of the innate immune response [33]. Moreover, these observations are valid in both macrophages and dendritic cells.
The IL-33 ST2 receptor is expressed on immune cells such as Th2, Treg, or even ILC2. AS of ST2 generates three isoforms: a transmembrane isoform named ST2L, a soluble isoform sST2, and ST2V [34]. sST2 acts as a “bait receptor” to competitively bind to IL-33 and block activation of the IL-33/ST2 signal [35][36][35,36] which plays an important role in the development of colorectal cancer [37].
The NLRP3 protein (NOD-like receptor family, pyrin domain containing 3) is involved in the NLRP3 inflammasome also composed of ASC (apoptosis-associated Speck-like protein containing CARD) and caspase-1. Largely expressed in macrophages, NLRP3 belongs to the PRR family (pattern recognition receptor), receptors for detecting molecules associated with pathogens. Upon activation, NLRP3 inflammasome cleaves pro-IL-1β and pro-IL-18 into IL-1β and IL-18, pro-inflammatory cytokines. NLRP3 is composed of three functional domains: PYD (N-terminal pyrin domain), the NACHT domain, and the LRR domain (leucine-rich repeat). LLR domain is responsible for inflammasome formation. A recent study has shown the existence of several alternative isoforms of NLRP3 in humans, losing either exon 5 or exon 7 or both exons [38]. The exclusion of exon 5 directly affects the LRR domain of NLRP3 that is involved in protein-protein interactions. As a result, the LRRΔE5 domain no longer interacts with the NEK7 protein (NIMA (never in mitosis A)—related kinase 7) [39]. As a result, caspase 1 is no longer able to convert pro-IL-1β and pro-IL-18. Thus, using antisense oligonucleotides, it would be possible to direct AS towards exon 5 exclusion in order to limit the onset of inflammation [38].
One of the first genes undergoing an AS to be identified in lymphocytes was Ptprc which encodes the CD45 protein. This tyrosine phosphatase is found in abundance on the surface of T and B lymphocytes, representing about 10% of total membrane proteins. CD45 is known to regulate TCR signaling by dephosphorylating Lck kinase. The pre-mRNA of Ptprc has three variable exons (4, 5, and 6) that generate the CD45RA, RB, RC, RO isoforms. In naïve T cells, CD45 is expressed either with variable exon 4 or with variable exon 5, namely CD45RA and CD45RB. In contrast, it was observed that in memory T cells all exons were excluded to form CD45RO [40]. There are functional differences between CD45 isoforms. Isoforms with a variable exon decrease the TCR activation threshold making cells more easily to be activated. The CD45RO isoform without variable exons attenuates TCR activation [41]. CD44 plays an important role in the migration of lymphocytes, in particular by allowing the phenomenon of rolling adherence and then firm adhesion with vascular endothelial cells expressing HA [42]. Alternative isoforms of CD44 have been observed in activated lymphocytes. The most expressed of these includes variable exon 5 (see CD44 part in cancer), and its appearance depends on the Sam68 RNA binding protein [43]. Sam68 has been shown to be activated by the Ras/Raf/MEK/ERK signaling cascade following the commitment of the TCR. Once active, Sam68 forms a complex with the SR SRm160 protein that interacts with the ESE sequence of exon 5 that induces its inclusion. This interaction with the CD44 pre-mRNA facilitates spliceosome recognition of splice sites. It has also been shown that by associating with the Brm protein, Sam68 is able to slow down the progression of RNA polymerase II that induces the inclusion of exon 5. Low splice sites are less attractive through their sequence for the spliceosome that prefers to attach to strong splice sites. This slowdown thus allows the spliceosome players to attach to the so-called weak splice sites surrounding exon 5. For the moment, little is known about the potential action of this alternative isoform in the immune response. Nevertheless, it is known that this isoform including exon 5 is more expressed in some autoimmune diseases [44].
3.3. Transcription Factors
The transcription factor Foxp3 is of great importance in regulatory T cells (Treg). In total, three isoforms of Foxp3 have been described: a full-length isoform Foxp3fl, another losing exon 2 Foxp3Δ2, and finally a last losing both exon 2 and exon 7, Foxp3Δ2Δ7. The different isoforms of Foxp3 do not all have the same effect on Treg differentiation. Indeed, while the first two maintain the immunosuppressive character of Treg lymphocytes, the latter does not. It has also been shown that when Foxp3fl and Foxp3Δ2Δ7 were coexpressed in comparable proportions, Foxp3Δ2Δ7 acts as a dominant negative and inhibits the activity of Foxp3fl [45]. It was also observed that exon 7 is of great importance for Foxp3. In individuals mutated at the donor splice site of intron 7, exon 7 is excluded. This results in a severe disruption of the immune system leading to IPEX syndrome (Immunodysregulation polyendocrinopathy enteropathy X-linked) manifested by polyendocrinopathy and autoimmune enteropathy [46]. Another study also described the impact of AS of the Foxp3 transcription factor in the plasticity of Treg cells. They first observed a significant increase in the Foxp3Δ2Δ7 alternative isoform in peripheral blood mononuclear cells (PBMC) and in intestinal biopsies of patients with Crohn’s disease compared to healthy patients. They then found that Tregs exposed to the pro-inflammatory cytokine IL-1β express Foxp3Δ2Δ7. However, the mechanism explaining the increased exclusion of the two Foxp3 variable exons is not yet explained. It is possible that this is due to the activation of splicing factors in the signaling pathway under the IL-1β receptor. Cells that express more this alternative isoform secrete more IL-17a, the cytokine characteristic of Th17 differentiation [45]. Indeed, this alternative isoform, unlike Foxp3fl, is not able to bind to the transcription factor RORγt to prevent it from inducing transcription of the gene encoding IL-17a. Thus, the Foxp3Δ2Δ7 isoform has a double effect in Crohn’s disease: inflammation is increased by the decrease in the Treg population but also by the increase in the Th17 population largely implicated in this disease [47].
Although Tbet is the key transcription factor in the differentiation of CD4 T subtype Th1, other transcription factors have been shown to be essential during the differentiation process. In Th1 cells, expression of IRF1 depends on either STAT1 via IFNγ or STAT4 via IL-12 pathways. This transcription factor has been described as essential for the expression of the IL-12 receptor β1 subunit [48][49][48,49]. Indeed, by acting in cooperation with Tbet, IRF1 allows the expression of a functional receptor provided with β1 and β2 subunits. This results in the amplification of the IL-12 cytokine signaling which supports Th1 lymphocyte orientation. There is an AS of Irf1 pre-mRNA in Th1 cells in vitro and in vivo under the influence of TGF-β. In fact, a short form of IRF1, lacking exon 7, appears during Th1 differentiation and its expression is increased in tumor settings and during exposure to TGF-β. A repressive action of IRF1-short in Th1 lymphocyte differentiation has been described. By interacting with the complete isoform of IRF1, IRF1-short prevents its binding to the Il12rb1 promoter [50].
3.4. Immune Checkpoints
PD-1 is encoded by the PDCD1 gene. The main PDCD1 AS event is exon 3 skipping, which results in the formation of the PD-1Δ3 isoform. PD-1Δ3 is a soluble form of PD-1 which prevents the interaction between PD-L1 and PD-1. Clinical studies have shown a correlation between prognosis and expression of PD-1Δ3 in patients with non-small lung cancer during treatment with erlotinib [51]. An antisense oligonucleotide targeting strategy can modify PDCD1 splicing and favor the generation of PD-1Δ3 to exert a therapeutic effect [52].
CTLA4 is another immune checkpoint that also has splice isoforms. Soluble CTLA4 (sCTLA4) inhibits the interaction between B7 and CD28 by binding to B7on antigen presenting cells. It thus inhibits the activation of T cells. Anti-sCTLA4 monoclonal antibodies bind specifically to sCTLA4 but do not recognize CTLA4, thus enhancing the T cell-specific response and exerting anti-tumor activity [53].