The sarcoplasmic reticulum (SR) is a specialized form of the endoplasmic reticulum of muscle cells, dedicated to calcium ion (Ca2+) handling, necessary for muscle contraction and relaxation.
- muscle
- Ca2+
- myopathy
- intracellular membrane
1. Introduction
sAnk1.5 and Obscurin Stabilize the SR around the Myofibrils
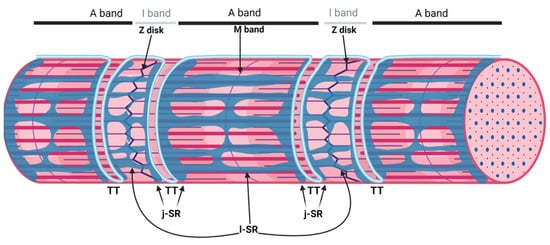
2. The Triad, a Unique Membrane System of the Skeletal Muscle
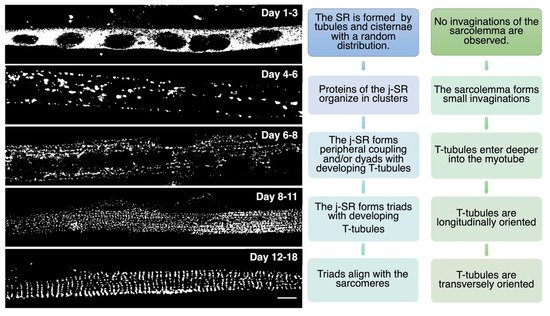
Triad Biogenesis, Repair, and Maintenance
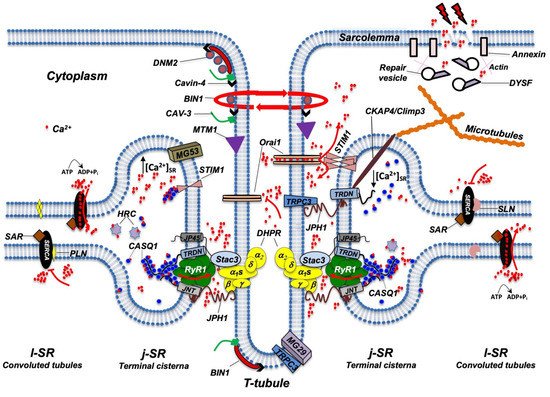
3. The Protein Complex of the ECC
At triads, several proteins participate in the ECC process, including integral membrane proteins such as RyR1, DHPR, triadin, junctin, j-SR protein 1 (jp45), and mitsugumin-56, the STAC3 adaptor protein, or luminal proteins such as the Ca2+ binding proteins calsequestrin (CASQ) and histidine-rich calcium (HRC) binding protein [14,105–107] (Figure 3).
3.1. RyR1, DHPR, and STAC3 Are Essential for ECC
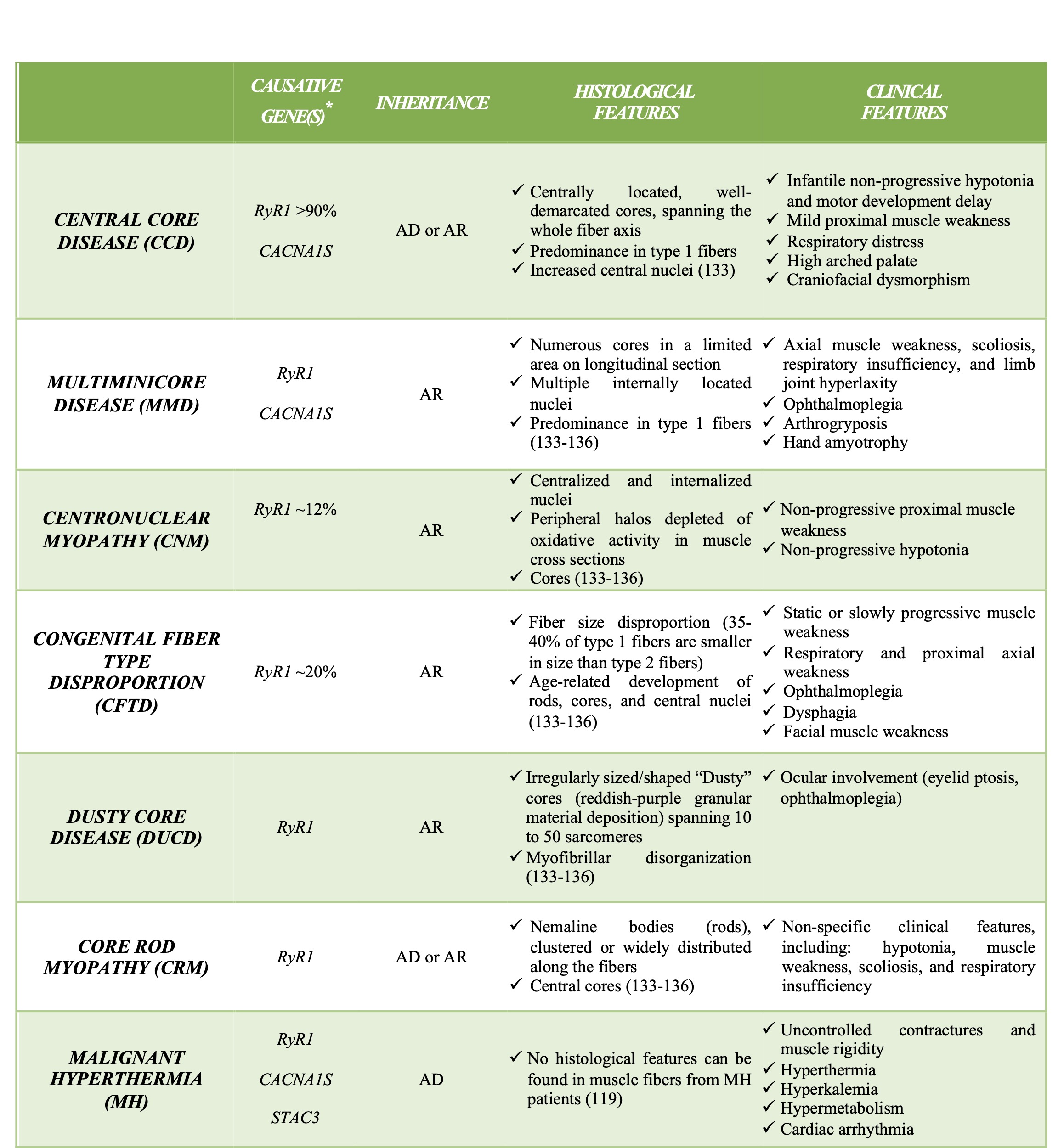
RyRs are a family of Ca2+ release channels that represent the largest ion channels known to date. In skeletal muscle, two RyR isoforms, RyR1 and RyR3, are expressed, although only RyR1 is essential for ECC activation. In skeletal muscle, RyR1 is primarily activated by a conformational coupling with the DHPR, a mechanism defined as depolarization-induced Ca2+ release (DICR) [13]. At variance with RyR1 channels, RyR3 channels are mainly expressed in neonatal versus adult muscles and are activated by a Ca2+-induced Ca2+-release (CICR) mechanism, whereby an increase in cytosolic Ca2+ causes RyR3 opening and, thus, Ca2+ release from intracellular stores [13,108,109]. RyR2 channels are mainly expressed in cardiac muscle, where they are involved in cardiac ECC through a CICR mechanism [105]. RyRs have a tetrameric structure, made by the assembly of four identical monomers, each consisting of a large N-terminal region of about 4300 amino acids extending in the sarcoplasm, while the remaining region contains the six transmembrane helices that anchor each monomer to the SR membrane and contribute to forming the channel pore region, followed by a short cytoplasmic C-terminal tail [110,111]. EM reconstruction studies have shown that RyRs display a mushroom-like form, with the cap in the cytoplasm representing 80% of the volume, and the stalk anchored in the j-SR membrane. In the cytoplasmic region, many domains have been described, referred to as subregions, that represent binding sites for several auxiliary proteins and molecules that contribute to regulating the opening and closing of the channel [110–112]. Calmodulin (CaM) was proposed to bind at different regions of RyR1 channels, acting either as a weak activator, at nanomolar Ca2+ concentration, or, in the micromolar range, as a channel inhibitor [113]. One of the proposed binding sites for CaM also binds the EF-hand protein S100, a protein also able to activate the channel [114]. RyR1 is also regulated by a member of the FK506-binding protein family, namely, FKBP12, which stabilizes the channel in a closed state [110,111,115,116]. Additional physiological or pharmacological RyR1 regulators include Mg2+, Homer 1c, the scorpion venom imperatoxin, halothane, dantrolene, PCB95, ryanodine, ruthenium red, methylxanthines, and 4-chloro-m-cresol [111,117,118]. Among these, dantrolene deserves special attention since it is now the only available agent for the treatment of malignant hyperthermia (MH), a pharmacogenetic disorder that results in a hypermetabolic state following exposure to volatile anesthetics or succinylcholine. MH is mainly associated with dominant mutations in human RYR1 that result in RyR1 hyperactivation, leading to massive and uncontrolled Ca2+ efflux from the SR. Dantrolene was historically used as a skeletal muscle relaxant, and later, it was demonstrated to be able to block calcium release from the SR. Although the molecular mechanism responsible for dantrolene action on RYR1 is still not completely defined, its introduction for the clinical treatment of MH led to a significant decrease in mortality, from more than 90% to less than 5% [119]. Finally, RyRs can also be regulated by post-translational modification, such as oxidation, nitrosylation, and phosphorylation/dephosphorylation cycles that occur via several kinases, such as PKA, PKG, Ca2+/CaM-dependent protein kinase II (CaMKII), and phosphatases (PP1, PP2A, and PDE4D3) [111,120].
DHPRs are L-type voltage-gated Ca2+ channels located on the TT. The skeletal muscle DHPR is composed of a heteromultimeric complex that includes the α1s, α2, and δ, β1α, and γ1 subunits [121]. The α1s subunit (also referred to as CaV1.1) is an integral membrane protein containing four transmembrane domains, each composed of six alpha helices, acting as the pore-forming and the voltage-sensing unit. The voltage-sensing domain of α1s interacts with the γ1 subunit that regulates channel inactivation [122]. The β1a subunit is also located in proximity to the voltage-sensing domain of the α1s subunit and supports the assembly of DHPRs in tetrads, where one DHPR tetrad faces one of the four subunits of the tetrameric RyR1 channel [121,123,124]. Mice knockout for either α1s or β1a subunits die perinatally, indicating that these subunits are essential for ECC [125,126]. The α2-δ subunits associate through four disulfide bonds and interact with the extracellular loops of the α1s subunit [121]. Despite its function as a voltage-gated calcium channel, the role of DHPR in skeletal muscle ECC is to induce Ca2+ release from the SR by physically interacting with RyR1, according to “orthograde” signaling [127]. RyR1 is also able to regulate DHPR activity by the so-called "retrograde" coupling effect [17,128]. In particular, it was demonstrated that, in the absence of RyR1, the Ca2+ current mediated by DHPR is smaller than that measured in the presence of RyR1, suggesting that RyR1 can promote the Ca2+ channel activity of DHPR and accelerates DHPR activation. Interestingly, this activity is unique to RyR1 since RyR2 was found not to be able to restore either orthograde or retrograde signaling. Different regions in DHPR and RyR1 have been proposed to be important for their reciprocal association and regulation; the II-III loop of the α1s subunit is generally accepted to be involved in both orthograde and retrograde coupling, while the site of interaction on RyR1 has not yet been completely defined [14,127,129].
STAC3 is a recently identified component of the ECC machinery. It is an adaptor protein that supports the trafficking of the α1s subunit of DHPR and regulates the coupling of DHPR with RyR1 [129–132]. STAC3 knockout mice present severe defects in muscle development, mass, and morphology; they are completely paralyzed and die shortly after birth [132]. Mutations in RYR1, DHPR, and STAC3 were identified in several human congenital myopathies [119,133–136], see Table 1.
3.2. Triadin and Junctin (JNT), Two Integral Membrane Proteins That Regulate ECC
Triadin is encoded by the TRDN gene that generates, via alternative splicing, four different isoforms named according to their molecular weight: the skeletal muscle isoforms are Trisk 95 and Trisk 51, both displaying a triadic localization; the main cardiac isoform Trisk 32 (also known as CT1) and Trisk 49, which does not localize at the j-SR in skeletal muscle fibers [137,138]. Triadin isoforms share a common cytoplasmic N-terminal and transmembrane domain but differ in the length and composition of their luminal C-terminal segment [139]. Trisk 95 contains two cysteine residues in its luminal domain, C270 and C649, that enable self-multimerization and is targeted at triads by specific domains localized both in the cytoplasmic and the luminal regions [140,141]. Triadin acts as a functional regulator of ECC by interacting with RyRs [142,143], junctin [144,145], calsequestrin [141,146–148], and the histidine-rich Ca2+-binding protein (HRC) [149]. Triadin may also play a structural role in supporting triad architecture by interacting with the microtubule-binding protein Climp-63, also known as Cytoskeleton-associated protein 4 (CKAP4) [150]. Triadin knockout mice present both ECC alterations and abnormal triads [151,152]. In contrast to heart muscle, where mutations in triadin are associated with heart disease, no skeletal muscle disease has been associated with triadin so far.
Junctin is structurally similar to triadin and, like triadin, can bind calsequestrin and the ryanodine receptor [144,145,153–155]. Junctin knockout mice exhibited increased contractility and Ca2+-cycling parameters in cardiac muscle [156], but no significant changes were observed in skeletal muscle [157]. Similarly, acute junctin downregulation in cardiomyocytes resulted in a significant increase in contraction [158], while in skeletal myotubes, it resulted in a decrease in Ca2+ release, suggesting that junctin may display a different role in cardiac and skeletal muscles [159].
3.3. Calsequestrin- and Histidine-Rich Calcium-Binding Protein Store Ca2+ in the SR Lumen
In skeletal muscle, two isoforms of calsequestrin (CASQ) have been identified: CASQ1 and CASQ2. CASQ1 is expressed in fast- and slow-twitch skeletal muscle fibers, whereas CASQ2 is expressed in slow-twitch skeletal muscle fibers and in cardiac muscle [160]. The two isoforms present a high sequence homology and basically only differ in their acidic C-terminus [161]. CASQs are intra-luminal SR soluble proteins with high capacity and low-affinity Ca2+-binding properties. [162,163]. CASQ1 exists as either a monomer or polymers, whose assembly depends on the SR Ca2+ concentration [164,165]. CASQ1 is formed by three negative thioredoxin-like domains surrounding a hydrophilic core [166]. Park and collaborators proposed a model where, when the ionic strength in the lumen of the SR increases, these domains fold together, and CASQ monomers start to polymerize. Following a further increase in Ca2+ concentration, front-to-front and back-to-back interactions take place. This finally results in the assembly of large ribbon-like structures, where negatively charged cavities may accommodate additional Ca2+ [167]. CASQ1 polymerization is a reversible process depending on the luminal Ca2+ concentration and the presence of CASQ-binding proteins or post-translational modification [168–171]. During sustained SR Ca2+ depletion, it was shown that almost all CASQ is present in its depolymerized status. In this condition, progressive closure of the RyRs was reported, with a consequent reduction in Ca2+release aimed to prevent dangerous levels of SR Ca2+ depletion. Accordingly, it has been proposed that CASQ1 depolymerization may represent the intracellular switch that induces RyR1 closing [172].
In skeletal muscle, CASQs interact with RyR1, triadin, and junctin, forming a quaternary Ca2+ release complex [144,145,148,173,174]. However, how these interactions regulate ECC still remains to be defined, since different studies report that CASQs may either inhibit or activate or even have no effect on the opening of RyRs [169,174–181]. CASQ1 knockout mice show mild atrophy, narrower terminal cisternae, and proliferation of multilayered junctions, as well as mitochondria alterations. They exhibit significantly reduced SR Ca2+ content and marked SR Ca2+ depletion during high-frequency stimulation [182,183]. Recently, it has been demonstrated that CASQ1 acts as a regulator of SOCE by binding to STIM1. In particular, CASQ1 overexpression was found to inhibit SOCE by reducing STIM1 clustering and STIM1/OraiI interaction [184,185]. Indeed, in CASQ1 knockout mice, SOCE is constitutively active, resulting in enhanced Ca2+ entry that may compensate for the reduced total SR Ca2+ content [186,187]. Mutations in the CASQ1 gene have been identified in patients affected by a rare vacuolar myopathy [188] and in patients with tubular Aggregate Myopathy, TAM [189,190].
HRC is far less abundant than CASQs, suggesting that it works as a secondary calcium-binding protein [191,192]. It is composed of a conserved N-terminal domain, a C-terminal cysteine-rich region, and a central histidine-rich region, which allow HRC to bind Ca2+ and to interact with triadin [193,194]. Under physiological Ca2+ levels, HRC has a pentameric structure, but when Ca2+-binding sites are saturated due to increased SR Ca2+concentration, the protein shifts to a monomeric form [195]. Although considered as a secondary calcium-binding protein, HRC appears to play a non-secondary role in regulating Ca2+ homeostasis. Indeed, HRC may function as a negative regulator of RyRs and, in cardiac muscle, it was suggested to interact with SERCA pumps, acting as a negative regulator of Ca2+ re-uptake [149,192,196,197].
4. SERCA Pumps at the l-SR Are Responsible for Ca2+ Re-Uptake from the Sarcoplasm
The l-SR is mainly involved in Ca2+ re-uptake, although some ligand-gated Ca2+ channels, such as inositol 1,4,5-trisphosphate receptors (InsP3R), which release Ca2+ to regulate signaling pathways apart from myofibrils contraction, are also present here [198]. SERCA pumps play the most important role in SR Ca2+ replenishing. SERCAs belong to the P-type ATPases family, a large superfamily of integral membrane proteins that pump ions and lipids across cellular membranes using the energy derived from ATP hydrolysis. The P-type ATPases family is composed of five subfamilies, defined as P1–P5. SERCAs belong to the P2A-ATPases subfamily and are specific for Ca2+ transport, while P4-ATPases act as phospholipid flippases to maintain lipid asymmetry in a variety of membranes [199,200]. SERCA pumps can transport two Ca2+ ions for each hydrolyzed ATP. In vertebrates, SERCA pumps are encoded by three different genes, named ATP2A1-3. More than 10 protein variants are generated through alternative splicing occurring in the 3′-end of the main transcripts [201,202].
ATP2A1 encodes two major skeletal muscle proteins: SERCA1a and SERCA1b, highly expressed in fast-twitch skeletal muscle fibers. SERCA1b is expressed during neonatal stages and in regenerating muscles, while it is replaced by SERCA1a in adult skeletal muscles [203–205]. Four isoforms of SERCA2 (a–d) are generated by alternative splicing of the ATP2A2 gene. SERCA 2a is expressed in cardiac muscle, slow-twitch skeletal muscle fibers, and smooth muscle cells. SERCA2b is reported to be ubiquitously expressed, and it represents the main SERCA isoform in the brain [206–208]. mRNA of SERCA2c was found in epithelial, mesenchymal, and hematopoietic cell lines, as well as in primary human monocytes [206] and cardiac muscle, while SERCA 2d has been identified in skeletal muscle, although its role has not yet been fully elucidated [209]. ATP2A3 codes for six isoforms (SERCA3a-f), mostly identified in non-muscle cells, although SERCA3a, 3d, and 3f were also detected in cardiac muscle cells [209,210]. The primary structure of the different SERCA isoforms is highly conserved. Nevertheless, the SERCA variants differ in their enzymatic properties. For instance, SERCA1a displays a maximal activity that is two-fold higher than that of SERCA2a [211,212], and differences in Ca2+ affinities have also been reported between SERCA2b and SERCA3 [212]. SERCA activity is finely regulated by numerous small proteins. The first identified regulator of SERCA is Phospholamban (PLN), which exerts an inhibitory effect on SERCA in cardiac muscle [213–215] and, to a lesser extent, in smooth muscle and slow-twitch oxidative skeletal muscle fibers where, however, its functional role appears to be marginal [216–218]. PLN exists in two forms, monomeric and pentameric. The first is considered the “active” inhibitory form, while the pentameric state is considered the “inactive” storage form of PLN. PLN regulation occurs through one-to-one interaction with SERCA [218]. At high cytosolic Ca2+ concentrations, the inhibitory effect of PLN is mitigated due to Ca2+/calmodulin kinase (CaMKII)- and/or Protein Kinase A (PKA)-dependent phosphorylation [219–221]. A second inhibitor of SERCA in skeletal muscle and atria is represented by sarcolipin (SLN). In contrast to PLN, which inhibits SERCA activity by reducing its affinity for Ca2+, SLN uncouples ATP hydrolysis from Ca2+transport [222], thus promoting futile cycling, resulting in ATP consumption and increased thermogenesis. Indeed, SLN knockout mice do not show any visible phenotype but show compromised thermogenic capacity when exposed to cold [222,223]. This suggests that the role of SLN may be related to muscle functions linked to body metabolism and heat production rather than contraction [223]. Similar to the j-SR, the l-SR also contains a Ca2+ binding protein, namely, Sarcalumenin (SAR) [224], which colocalizes and interacts with SERCA2 [225]. Furthermore, SAR has been shown to play an essential role in maintaining cardiac function under biomechanical stresses and endurance exercise training [226,227]. Mutations in SERCA1 are associated with Brody disease, an extremely rare autosomal recessive myopathy (with a prevalence estimated at around 1 in 10 million births) characterized by muscle stiffness, myalgia and muscle cramps, aggravation of symptoms upon exposure to cold temperatures, and, in a smaller percentage of patients, muscle weakness. It is basically characterized by exercise-induced impairment of fast-twitch skeletal muscle relaxation due to diminished SERCA1 activity. This increased levels of cytosolic Ca2+ due to SERCA1 impairment directly contribute to muscle stiffness [228–230].
Although SERCAs represent the key enzymes that contribute to sarcoplasmic Ca2+ clearance following muscle contraction, a trans-sarcolemmal flux toward the extracellular environment also occurs in striated muscles. This is mediated by Plasma Membrane Calcium ATPases (PMCAs) and Na+/Ca2+ exchangers localized in the sarcolemma. Similar to SERCAs, PMCAs also belong to the P-type ATPase superfamily. They can transport one Ca2+ for each hydrolyzed ATP and are directly activated by interaction with Ca2+-calmodulin, which enhances PMCAs affinity for Ca2+ [231]. PMCAs are encoded by four different genes (ATP2B1-4) that, by alternative splicing, generate multiple isoforms [232]. Among these, PMCA3 and PMCA1 were found to be expressed in skeletal muscle, and PMCA1 was specifically localized on the T-tubule membrane [233,234]. NCX catalyzes the exchange of three Na+ ions and one Ca2+ ion across the sarcolemma in a high capacity and low Ca2+ affinity manner. Depending on the electrochemical gradient across the plasma membrane, NCX can either extrude intracellular Ca2+ (according to the so-called forward mechanism) or take up extracellular Ca2+ (a reverse mechanism) [235]. In mammals, three genes, NCX1, NCX2, and NCX3, are present; NCX3 is expressed in brain and adult skeletal muscle [236,237], while NCX1 is expressed in developing skeletal muscles and in cardiac muscle [237]. The role of NCX has been mainly defined in cardiac muscle cells, where it represents the major mechanism of Ca2+ extrusion from the cytoplasm during diastole [238,239].
5. Ca2+ Entry Units (CEU): Novel SR/Plasma Membrane Contact Sites to Refill Intracellular Ca2+ Stores
Entry of Ca2+ from the extracellular environment in response to a decrease in the endoplasmic reticulum Ca2+ content is a ubiquitous mechanism present in all cell types. The key elements of this pathway, named Store Operated Calcium Entry (SOCE), are STIM1 and Orai1 proteins [240]. STIM1 is a transmembrane protein localized in the ER membrane that acts as a sensor of luminal Ca2+ content; when intracellular Ca2+ stores are full, it is in a Ca2+-bound monomeric conformation. Depletion of the intracellular Ca2+ store results in dissociation of Ca2+ from STIM1, which forms aggregates that relocate at ER–plasma membrane contact sites, where they interact with and activate the Orai1 channels on the plasma membrane, allowing Ca2+ entry from the extracellular environment and refill of intracellular stores. Ca2+ entry from the extracellular environment through the SOCE mechanism is also present in skeletal muscle [241]. More recently, the requirement of extracellular Ca2+ to refill the SR Ca2+ stores, after intense, prolonged activity, has been shown to result in the assembly of additional intracellular junctions between the SR and the plasma membrane, providing additional space for STIM1 and Orai1 interaction. These newly identified sites, named Ca2+ Entry Units (CEU), support increased Ca2+ entry via Orai1, and their activity appears to contribute to improving fatigue resistance under continuous muscle activity [242–244] (Figure 4). Mutations in Orai1 and STIM1 have been identified in patients with tubular aggregate myopathy (TAM), a rare genetic disease characterized by accumulation, in type-2 fibers, of highly ordered membrane tubules containing SR proteins, such as CASQ1, SERCA, triadin, RyR1, and STIM1 [245–249]. More recently, mutations in CASQ1 have also been identified in TAM patients [189,190].
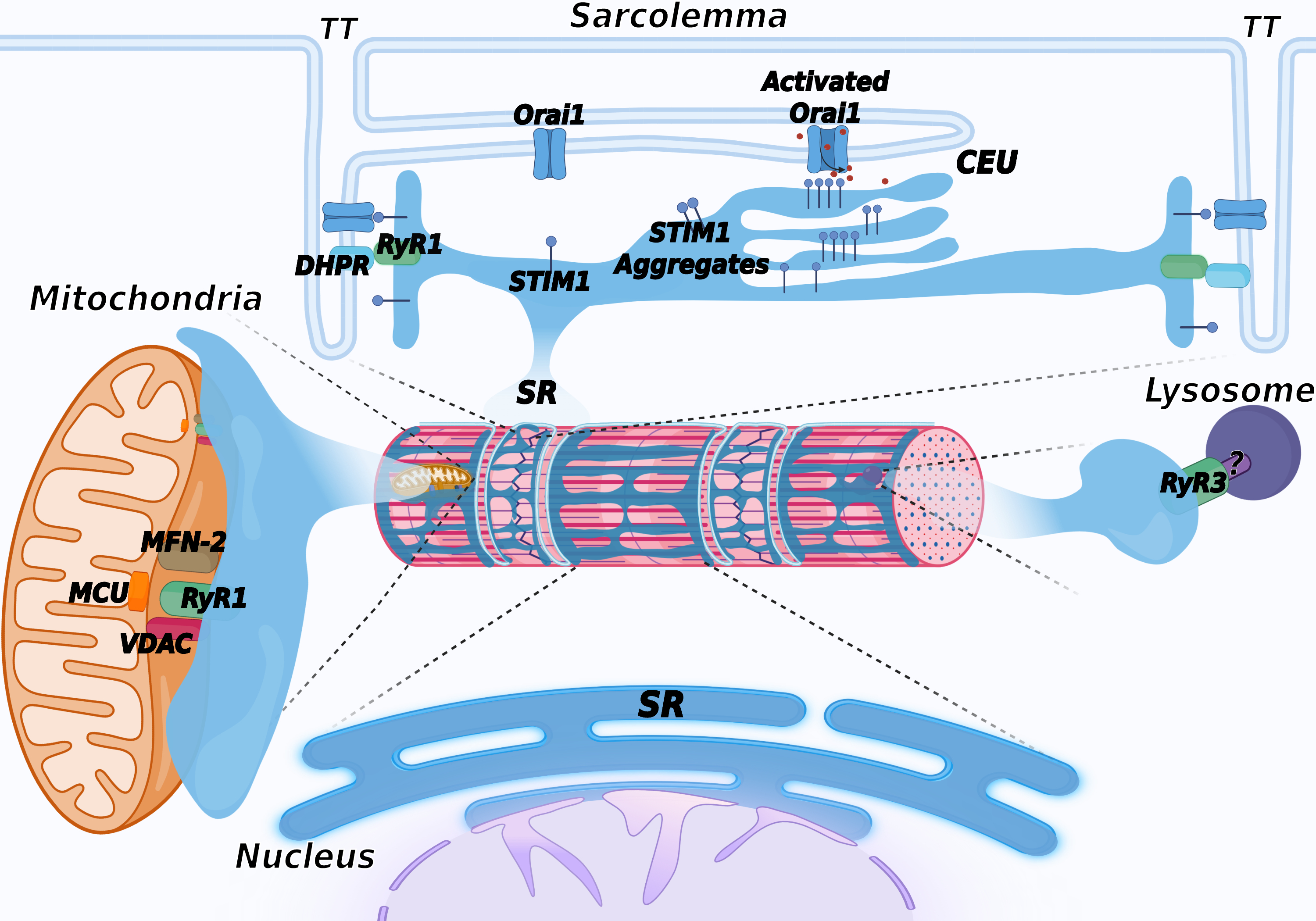
Figure 4. Schematic representation of membrane contact sites contributed by the SR. In addition to triads, the SR contributes to the formation of additional membrane contact sites in muscle cells. Depletion of intracellular Ca2+ stores in the SR induces activation of SOCE, mediated by a physical interaction between STIM1, a Ca2+ sensor of the SR and Orai1, a Ca2+ channel located in TT, allowing entry of Ca2+ from the extracellular space. Repetitive stimulation of muscle contraction was found to promote SR and TT remodeling to form additional sites of interaction between STIM1 and Orai1, called calcium entry units (CEU). These are formed by stacks of flat cisternae of the SR that make contact with elongated tubules extending from TT. In striated muscles, mitochondria are mostly positioned adjacent to triads. Association with the SR is mediated by the voltage-dependent anion channel 1 (VDAC) and RyR. Mitofusin-2 (MFN-2) also contributes to tethering SR and mitochondria. These contact sites are aligned with the inner mitochondrial membrane, where the mitochondrial Ca2+ uniporter (MCU) is located. Lysosome–SR nanojunctions mediated by RyR3 channels have been first described in pulmonary arterial myocytes; the RyR3 interactor present on the lysosomal membrane is not known and is therefore indicated with a question mark (?). More recently, these junctions have also been observed in cardiac muscle, while no data are currently available concerning skeletal muscle. The outer nuclear membrane is continuous with the membrane of the SR, allowing the arrangement of a continuous Ca2+storage system between the SR and the nuclear envelope. The inner nuclear membrane forms invaginations that enter the nuclear matrix to support intra-nuclear Ca2+ signaling. Created with BioRender.com. accessed on 14 march 2022
6. Mitochondria-Associated Membranes (MAM)
Interaction between the ER/SR and mitochondria occurs at specific membrane contact sites, generally defined as Mitochondria Associated Membranes (MAMs), populated by several proteins and protein complexes that act as tethers between the SR and the outer mitochondrial membrane [250]. In striated muscles, mitochondria are mostly positioned in the inter-myofibrillar spaces adjacent to triads, although contacts between mitochondria and the longitudinal SR have also been observed [251–254]. Association with the SR is mediated by the voltage-dependent anion channel 1 (VDAC) [255]. In cardiac muscle, it has been proposed that VDAC interacts with RyR2 [256]. Mitofusin-2 (MFN-2) also contributes to tethering SR and mitochondria in skeletal muscles [257,258]. Interestingly, during mouse skeletal muscle development, mitochondria redistribute from a longitudinal arrangement towards a triad-associated distribution, with an increase in the frequency of contacts until the first four months after birth [258]. These contacts are aligned with domains of the inner mitochondrial membrane enriched in the mitochondrial Ca2+ uniporter (MCU), a low-affinity Ca2+ transport protein (Figure 4). It has been shown that RyR1 opening results in the formation of local micro-domains, where Ca2+ concentration may be up to 10 folds higher than in the rest of the cytoplasm [259]. This high Ca2+ concentration allows the transfer of Ca2+ to the mitochondrial matrix through the uniporter [260]. From a functional point of view, increases in mitochondrial Ca2+ concentration regulate ATP production by enhancing the synthesis of NADH and FADH2 [261,262], thus linking ATP production to muscle demand. Accordingly, slow-twitch fibers in skeletal muscle that are highly oxidative have the highest mitochondrial content compared to fast-twitch fibers; as a result, MCU deletion results in impairment in muscle force and in oxidative metabolism [263]. In cardiac muscle, the MCU-mediated Ca2+ influx was shown to play an important role during the fight-or-flight response following catecholaminergic stimulation [264].
Alterations of SR/mitochondria interaction or of mitochondrial morphology and function in striated muscles are often observed in human muscle diseases or mouse models of muscle diseases characterized by alterations in Ca2+ signaling [251,265–267]. However, the exact mechanisms linking alterations of MAM, mitochondrial function, and muscle diseases are still to be defined.
7. The SR and the ER: Two Faces of a Single Organelle
This review started with the statement that the SR is a specialized form of ER of muscle cells dedicated to Ca2+ handling. One may therefore ask what happened to the ER in striated muscle cells. In eukaryotic cells, the ER is dedicated to Ca2+ storage, protein synthesis and folding, and lipid and sterol synthesis. Although ER-related functions are clearly present in striated muscles, the distribution of the ER within the SR membranes is less evident. The recognized idea is that the SR and the ER represent a continuous membrane system made of different specialized subdomains [268]. Compartmentalization of ER and SR markers was demonstrated in skeletal muscle fibers, where ER-specific proteins were detected at the perinuclear region and in two distinct rough ER sub-compartments: the first, in correspondence of the I band that does not apparently contain ER exit sites, and the second, near the Z disk, which sustains export activity towards the Golgi [105,269]. Similarly, in cardiac muscle cells, protein synthesis was found to occur not only around the nuclei but also within ER/SR membranes surrounding the sarcomeres; interestingly, translation of some SR membrane proteins has been proposed to occur starting from pools of mRNAs that are transported from the perinuclear region towards ER/SR protein-synthesis sites through a microtubule-dependent system [270]. A less-clear distinction can be observed in smooth muscle, where ER and SR markers are basically overlapping [271].
7.1. Additional Contact Sites Contributed by the ER/SR
In eukaryotic cells, the ER is engaged in a complex network of interactions with many intracellular components, such as the nuclear envelope (NE), the plasma membrane, mitochondria, the Golgi complex, peroxisomes, endosomes, lysosomes, and lipid droplets [272]. All these interactions have been less characterized in striated muscle cells. Lysosome–SR nanojunctions were described in pulmonary arterial myocytes (Figure 4). These junctions were identified between clusters of lysosomes and perinuclear regions of the SR rich in RyR3 and are believed to provide an intracellular structure involved in the regulation of specific Ca2+-signaling events. These junctions were identified based on the observation that nicotinic acid adenine dinucleotide phosphate (NAADP), a Ca2+-mobilizing second messenger, induced the release of Ca2+ from a lysosome-related Ca2+ store, and this was amplified by the generation of intracellular Ca2+ waves supported by RyR3 opening, following a CICR mechanism [273]. According to these studies, lysosomes act as a trigger zone through which Ca2+ is released with a quantal pattern; upon reaching an activation threshold, RyR3 channels open, and calcium oscillations are generated. More recently, nanojunctions between lysosomes, SR, and mitochondria have been described at the ultrastructural level in rabbit ventricular myocytes. At difference with junctions observed in pulmonary arterial myocytes, in cardiac myocytes, lysosomes are distributed with a periodicity consistent with the length of a sarcomere, allowing the association with specific areas of the SR [274]. The role of NAADP-mediated signaling in cardiac muscle cells is suggested to be involved in intracellular Ca2+ regulation, mainly following beta-adrenergic stimulation [275].
In striated muscle cells, stacks of ER/SR cisternae distribute in the perinuclear regions and connect with the nuclear envelope (NE) with functions correlated with protein synthesis and the regulation of intra-nuclear Ca2+signaling (Figure 4). The NE is formed by two phospholipid bilayers that form the inner nuclear membrane and the outer nuclear membrane, outlining an internal space called the perinuclear cisternae. The outer nuclear membrane is continuous with the membrane of the rough ER; this connection results in the formation of common luminal space with the ER, which, in muscle cells, allows the arrangement of a continuous Ca2+ storage system between the SR and the NE [276]. EM analysis showed the existence of membrane invaginations of the inner nuclear membrane that enter the nuclear matrix to support different functions, including intranuclear Ca2+signaling [277]. InsP3R, RyRs, and SERCA have been described at these sites; in striated muscle cells, Ca2+release is apparently mainly regulated by RyRs [278,279], while both InsP3R and RyRs are present in smooth muscle cells [280]. In cardiac myocytes, STIM1 and Orai1 have also been described in the inner nuclear membrane, suggesting that the intranuclear Ca2+ signaling may be more complex than previously expected [281].
An additional issue is represented by the SR/nuclei juxtaposition within the overall skeletal muscle architecture. In skeletal muscle, nuclei are positioned at the fiber periphery between myofibrils and the sarcolemma, in proximity to T-tubules. The positioning of nuclei is regulated by different mechanisms, including the formation of a specific microtubule network, and is critical for muscle function; indeed, improper myonuclear localization is observed in muscle diseases, such as centronuclear myopathy and muscular dystrophies [282]. The molecular link supporting nuclear juxtaposition with the SR is currently not known. A recent work performed in skeletal muscles of Drosophila melanogaster suggests that this interaction may be mediated by a three-partner connection where amphiphysin 2 on TT binds, at the same time, Ma2/d, a protein present on both TT and SR membranes, and, at the nuclear membrane, Msp30, a protein previously described to mediate connections between the NE and the Z disks [283–285].
8. Concluding Remarks
This review provides an updated view of the structural, molecular, and functional organization of the SR in skeletal muscle, highlighting some of the specialized domains whereby this organelle supports activation of muscle contraction. The historical knowledge that the terminal cisternae of the SR engage with the T-tubules in forming the triad, which represents the first identified membrane contact site, is now expanded by evidence that in skeletal muscle, the SR participates in the assembly of at least two more types of membrane-contact sites.
In one case, the identification of CEUs, dynamic membrane contact sites consisting of stacks of SR cisternae and the extension of T tubules, structurally and functionally different from triads, provides evidence of how skeletal muscle fibers can sustain refilling of intracellular Ca2+ stores under conditions of prolonged activity. The second contact site, MAM, between the SR and mitochondria, has the functional role of synchronizing the mitochondrial energy production rate with the metabolic demand of muscle fibers. Altogether, these new findings are changing the traditional view of the SR as a static organelle by depicting an unexpected dynamic ability to extend its complex structure to support muscle function.
At the same time, there are several points that remain to be clarified. A major open question relates to the precise positioning of triads at the boundary of the I and A bands of the sarcomere. This highly precise pattern, evident in EM micrographs for about a half-century, is waiting for the identification of the proteins that tether triads to the sarcomere with such a regularly repetitive precision. Concerning CEU, we need to identify the molecules that support the dynamic assembly and disassembly of CEU and how Orai1 and STIM1 are recruited at these sites. Mitochondria, like many other organelles, in skeletal muscle, are localized with a well-defined pattern with respect to triads and sarcolemma, a pattern that differs in slow- and fast-twitch fibers. Additionally, in this case, the molecules that anchor mitochondria to these domains of muscle fibers remain to be identified.
In conclusion, we can anticipate that the above list is, certainly, still missing additional SR domains that are yet to be identified; these might probably involve activities related to the regulation of Ca2+ homeostasis, but more likely, additional functions necessary to keep skeletal muscle fibers healthy and fully functional.