Artificial intelligence (AI) refers to machines, mainly computers, working like humans. In AI, machines execute tasks such as speech recognition, solving problems, and learning. Machines can work and act like humans if they have enough instruction and knowledge. Drug development is a costly and time-consuming business, and only a minority of approved drugs generate returns exceeding the research and development costs. As a result, there is a huge drive to make drug discovery cheaper and faster. With modern algorithms and hardware, it is not too surprising that the new technologies of artificial intelligence and other computational simulation tools can help drug developers.
- artificial intelligence
- machine learning
- drug design
- COVID-19
1. Introduction
Artificial intelligence (AI ) systems can be divided into four groups based on machines’ ability to use past experiences to predict future decisions: (1) Machine learning refers to AI where machines are not explicitly programmed to perform tasks, but they learn and improve from training automatically. Deep learning is a subdivision of machine learning based on artificial neural networks for predictive analysis. (2) Natural language processing (NLP) is the interaction between computers and human language. (3) Automation and robotics aim to allow monotonous and repetitive tasks to be performed by machines. (4) Machine vision—machines can capture visual information and then analyze it [7][1]. AI has been used in applications to solve specific problems throughout industry and academia. Like electricity or computers, AI is a general-purpose technology that has a multitude of applications. It has enhanced language translation, image recognition, credit scoring, e-commerce, and many other aspects of our lives.
In the field of computer-aided drug discovery (CADD), the scientific community worldwide is regularly developing new technologies and algorithms to obtain hit compounds in a short time and reduce the overall cost [8][2]. Nowadays, the introduction of artificial intelligence (AI) and all the related techniques such as deep learning (DL), machine learning (ML), and other classical computational chemistry tools to drug discovery has had a significant effect on the success rate and velocity of novel pharmaceutical identification [9][3].
AI and classical CADD tools can be used alone or combined to produce new approaches that integrate a broad range of algorithms with enhanced prediction capabilities. CADD is classically classified into two methods: structure-based drug design and ligand-based drug design. Both are faces of the same coin and massively rely on force fields, scoring functions, and algorithms to evaluate and rank the studied molecules’ energy contribution in the targeted macromolecular biological system. While computer-aided structure-based drug design (e.g., docking) depends on the actual 3D structure of the targeted binding site of the targeted receptor protein to understand the stabilizing interactions at the molecular level between the studied ligand/receptor system, the ligand-based drug design (e.g., 3D-QSAR modeling) approach relies on the recognition of a database of already-known ligands interacting with the target receptor. Both structure- and ligand-based technologies have several success stories and play key roles in the drug modern drug discovery process [10,11,12,13,14][4][5][6][7][8].
2. Structure-Based Artificial Intelligence Methods for Small Molecules
| n | Structure | CmDock Docking Score |
|---|
| 1 | 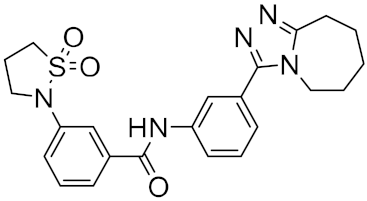 |
−32.51 |
| 2 | 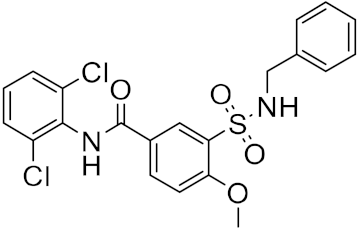 |
−29.02 |
| 3 | 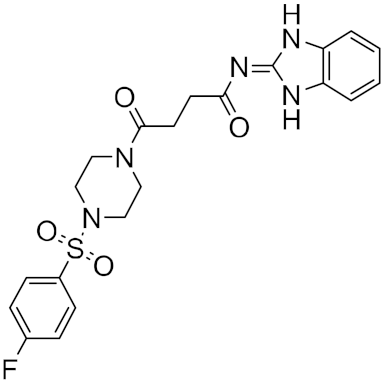 |
−26.80 |
| 4 | 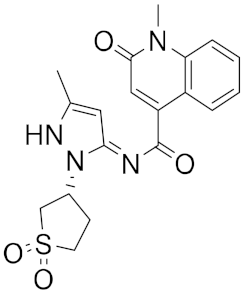 |
−25.58 |
| 5 | 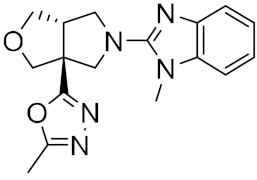 |
−25.53 |
| 6 | 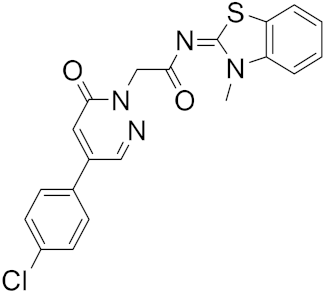 |
−25.05 |
| 7 | 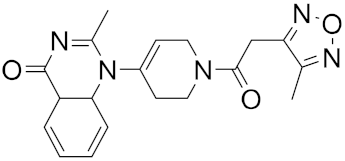 |
−24.76 |
| 8 | 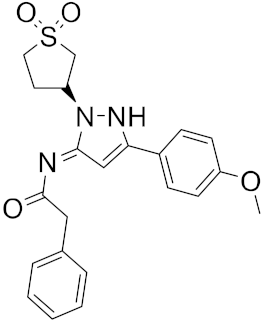 |
−24.51 |
| 9 | 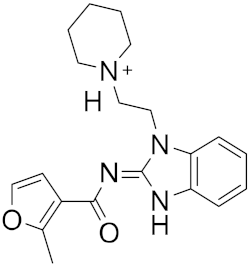 |
−24.17 |
| 12 | 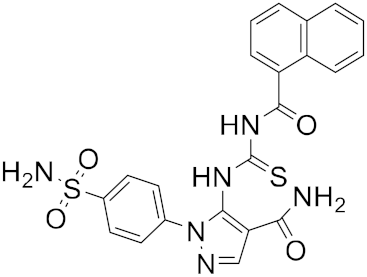 |
−24.01 |
| 11 | 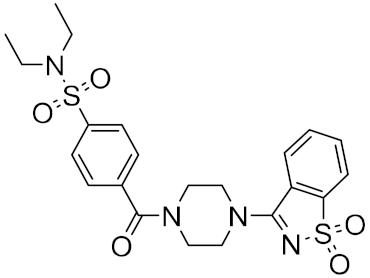 |
−23.98 |
| 12 | 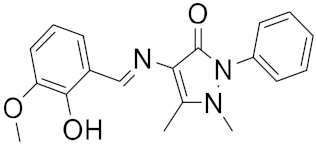 |
−23.61 |
| 13 | 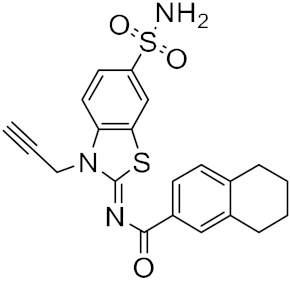 |
−23.53 |
| 14 | 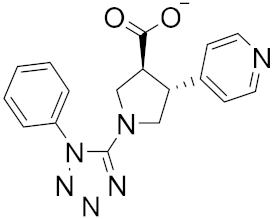 |
−23.26 |
| 15 | 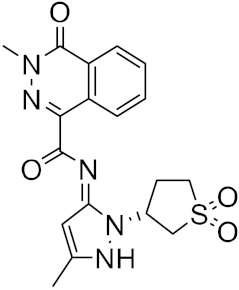 |
−23.18 |
A different research group screened a library of molecules for their potential ability to inhibit SARS-CoV-2’s main protease (Mpro) and the receptor-binding domain (RBD) of the spike protein by using the molecular docking software AutoDock Vina [27][20]. Mpro is a peculiar cysteine protease of the coronavirus family and has a crucial role in mediating viral replication and transcription. The absence of a homologous human protease makes this protein an important target against COVID-19 [28][21].
Recently, Born et al., devised a new method for discovering and synthesizing drugs against SARS-CoV-2 [30][22]. This procedure merges biology and chemistry for target-driven molecular design associated with an automatic synthesis plan generator and can be virtually applied to any protein target. The approach uses deep generative models that implement a conditional molecule generator to propose drug candidates by exploring the latent space of proteins and small molecules. The promise of this approach is the possibility to generalize to unseen targets. In this way, a conditional molecular generator can produce novel structures expressly designed to target a protein of interest [31][23]. The founders of COVID Moonshot, a non-profit, open-science consortium of scientists from around the world dedicated to discovering globally affordable and easily manufactured antiviral drugs against COVID-19, demonstrated the utility of a de novo design using machine learning with synthesis route prediction [33][24]. The purpose of the study was to generate potential new drugs targeting the main protease (Mpro) of the novel coronavirus. In fact, while classic approaches tend to modify existing compounds, exploring limited chemical space, this machine learning method searches in a larger chemical space. The inconvenience of this approach is the expense of synthetic accessibility, but this can be overcome using machine learning to predict the synthetic route. Pirolli D. et al., with the support of machine learning approaches, reported a structure-based virtual screening as an effective strategy to discover inhibitors of protein–protein interactions (PPIs) between SARS-CoV-2 RBD and human ACE2 using the ZINC database [42][25]. Using different ligand- and structure-based approaches, a customized virtual screening (VS) strategy was set up. The first step of the VS strategy was the selection of a library focused on small molecule PPIs from a dataset of 2 million compounds, using a ligand-based approach capable of recognizing chemical characteristics and scaffolds common to known modulators. For this purpose, a convolutional neural network (cNN) was trained to obtain a QSAR model capable of identifying potential PPI modulators within a virtual library of unknown molecules. The molecules classified by the cNN-based QSAR as potential PPI modulators were further filtered by the expected toxicological properties to discard compounds harmful to human health. The resulting virtual library was hooked to ACE2 to identify compounds with the best binding affinity for the spike protein interaction surface. The dataset of 30,029 ligands obtained from QSAR modeling and toxicity analysis against the druggable site-4 pocket region was used to screen for effective inhibitors of the protein–protein interactions of the SARS-CoV-2 RBD/ACE2 tip. Then a virtual Glide screening in SP mode was performed, and the next phase was done by XP docking. Based on the docking score, the first 15,015 classified ligands (50%) were selected and reassessed with Prime MM-GBSA to estimate their binding free energy. The compounds were then filtered based on distance constraints by selecting only small molecules within 4.5 Å away from any atom of Tyr83 and Gln24 residues. The remaining 9730 molecules were then grouped based on their diversity, and the resulting 973 virtual hit compounds were further assembled into 66 clusters using interaction fingerprints.3. Ligand-Based Artificial Intelligence Methods for Small Molecules
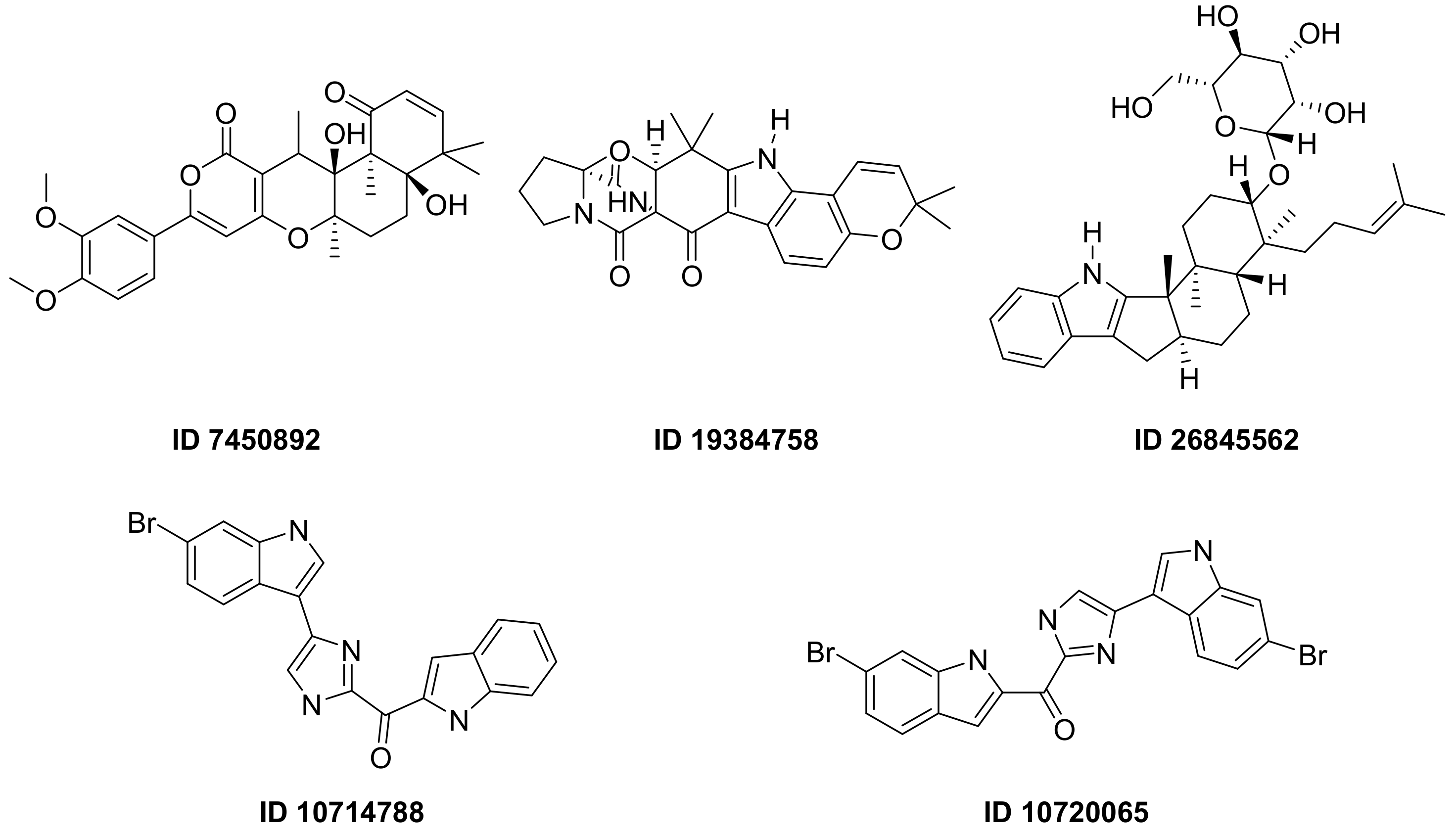
F. Pereira et al., succeeded in predicting five new inhibitors against SARS-CoV-2 Mpro using a CADD method based on a quantitative structure-activity relationship (QSAR) classification model that was built from 5276 organic molecules extracted from the ChEMBL database. Virtual screening was then performed using 11,162 marine natural products (MNPs) retrieved from the Reaxys® database. From the QSAR approach, 494 MNPs were selected and subsequently subjected to molecular docking against the Mpro. Among the evaluated compounds, five MNPs have been proposed as the most promising marine drugs as inhibitors of SARS-CoV-2 Mpro, among them a benzo[f]pyrano [4,3-b]chromene (Reaxys ID 7450892), notoamide I (Reaxys ID 19384758), hemindole SB beta-mannoside (Reaxys ID 26845562), and two derivatives of bromoindole (Reaxys IDs 10,714,788 and 10720065) Figure 21 [43][26].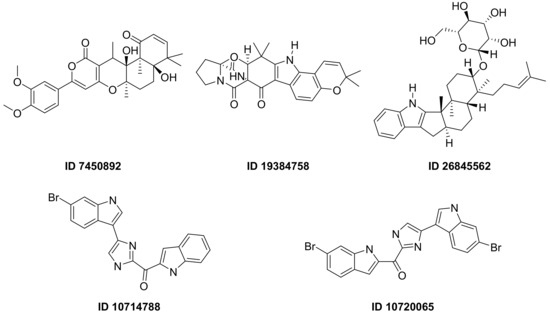
4. Artificial Intelligence Methods Vaccine Design
References
- Glikson, E.; Woolley, A.W. Human Trust in Artificial Intelligence: Review of Empirical Research. Acad. Manag. Ann. 2020, 14, 627–660.
- Yu, W.; MacKerell, A.D., Jr. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 85–106.
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A., Jr.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364.
- Floresta, G.; Apirakkan, O.; Rescifina, A.; Abbate, V. Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis. Molecules 2018, 23, 2183.
- Floresta, G.; Abbate, V. Machine learning vs. field 3D-QSAR models for serotonin 2A receptor psychoactive substances identification. RSC Adv. 2021, 11, 14587–14595.
- Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Mar. Drugs 2018, 16, 384.
- Floresta, G.; Cilibrizzi, A.; Abbate, V.; Spampinato, A.; Zagni, C.; Rescifina, A. 3D-QSAR assisted identification of FABP4 inhibitors: An effective scaffold hopping analysis/QSAR evaluation. Bioorg. Chem. 2019, 84, 276–284.
- Floresta, G.; Gentile, D.; Perrini, G.; Patamia, V.; Rescifina, A. Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach. Mar. Drugs 2019, 17, 624.
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology 2001, 279, 371–374.
- Srinivasan, S.; Batra, R.; Chan, H.; Kamath, G.; Cherukara, M.J.; Sankaranarayanan, S. Artificial Intelligence-Guided De Novo Molecular Design Targeting COVID-19. ACS Omega 2021, 6, 12557–12566.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461.
- Mohapatra, S.; Nath, P.; Chatterjee, M.; Das, N.; Kalita, D.; Roy, P.; Satapathi, S. Repurposing therapeutics for COVID-19: Rapid prediction of commercially available drugs through machine learning and docking. PLoS ONE 2020, 15, e0241543.
- Verma, N.; Qu, X.; Trozzi, F.; Elsaied, M.; Karki, N.; Tao, Y.; Zoltowski, B.; Larson, E.C.; Kraka, E. SSnet: A Deep Learning Approach for Protein-Ligand Interaction Prediction. Int. J. Mol. Sci. 2021, 22, 1392.
- Karki, N.; Verma, N.; Trozzi, F.; Tao, P.; Kraka, E.; Zoltowski, B. Predicting Potential SARS-CoV-2 Drugs-In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking. Int. J. Mol. Sci. 2021, 22, 1573.
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904.
- Jukic, M.; Skrlj, B.; Tomsic, G.; Plesko, S.; Podlipnik, C.; Bren, U. Prioritisation of Compounds for 3CL(pro) Inhibitor Development on SARS-CoV-2 Variants. Molecules 2021, 26, 3003.
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337.
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412.
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107.
- Kwofie, S.K.; Broni, E.; Asiedu, S.O.; Kwarko, G.B.; Dankwa, B.; Enninful, K.S.; Tiburu, E.K.; Wilson, M.D. Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules 2021, 26, 406.
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 M(pro): A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607.
- Born, J.; Manica, M.; Cadow, J.; Markert, G.; Mill, N.A.; Filipavicius, M.; Janakarajan, N.; Cardinale, A.; Laino, T.; Martinez, M.R. Data-driven molecular design for discovery and synthesis of novel ligands: A case study on SARS-CoV-2. Mach. Learn.-Sci. Technol. 2021, 2, 025024.
- Meyers, J.; Fabian, B.; Brown, N. De novo molecular design and generative models. Drug Discov. Today 2021, 26, 2707–2715.
- Morris, A.; McCorkindale, W.; Consortium, T.C.M.; Drayman, N.; Chodera, J.D.; Tay, S.; London, N.; Lee, A.A. Discovery of SARS-CoV-2 main protease inhibitors using a synthesis-directed de novo design model. Chem. Commun. 2021, 57, 5909–5912.
- Pirolli, D.; Righino, B.; De Rosa, M.C. Targeting SARS-CoV-2 Spike Protein/ACE2 Protein-Protein Interactions: A Computational Study. Mol. Inform. 2021, 40, e2060080.
- Gaudencio, S.P.; Pereira, F. A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition. Mar. Drugs 2020, 18, 633.
- Yassine, R.; Makrem, M.; Farhat, F. Active Learning and the Potential of Neural Networks Accelerate Molecular Screening for the Design of a New Molecule Effective against SARS-CoV-2. Biomed. Res. Int. 2021, 2021, 6696012.
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2021, 22, bbaa161.
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 2021, 11, 3238.
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, 2680160.
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29.
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS ONE 2012, 7, e45152.
- Saha, S.; Raghava, G.P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48.
- El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. 2008, 21, 243–255.
- Li, M.; Jiang, Y.; Gong, T.; Zhang, Z.; Sun, X. Intranasal Vaccination against HIV-1 with Adenoviral Vector-Based Nanocomplex Using Synthetic TLR-4 Agonist Peptide as Adjuvant. Mol. Pharm. 2016, 13, 885–894.
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Roder, G.; Peters, B.; Sette, A.; Lund, O.; et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2007, 2, e796.
- Emini, E.A.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839.
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Peters, B.; Sette, A.; Justesen, S.; Buus, S.; Lund, O. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput. Biol. 2008, 4, e1000107.
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J. Proteome Res. 2020, 19, 2304–2315.
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388.