The existence of orderly structures such as tissues and organs is made possible by cell adhesion, i.e. the process by which cells attach to neighbouring cells and a supporting substance in the form of the extracellular matrix. The extracellular matrix is a three-dimensional structure composed of collagens, elastin and various proteoglycans and glycoproteins. It is a storehouse for multiple signalling factors. Tissue disruption often prevents the natural reconstitution of the matrix. The use of appropriate implants is then required. The possibilities of regenerating damaged tissues using an artificial matrix substitute are described, detailing the host response to the implant. An important issue is the surface properties of such an implant and the possibilities of their modification.
- extracellular matrix
- cellular receptors
- cell adhesion
- cell signalling
- scaffolds
- biomaterials
1. The Extracellular Matrix—Composition, Structure, Functions
1.1. Two Types of the Extracellular Matrix
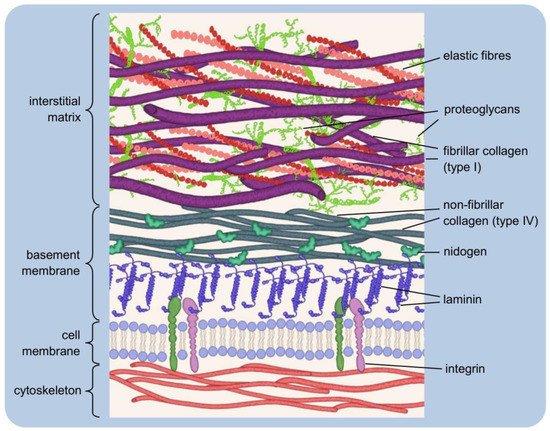
1.2. Major Components of the Extracellular Matrix and Their Functions
1.2.1. Collagens
1.2.2. Elastin
1.2.3. Proteoglycans
1.2.4. Glycoproteins
1.3. The Dynamic Structure of the Extracellular Matrix
1.4. The Extracellular Matrix as a Storehouse of Growth Factors
2. Artificial Substitutes of the Extracellular Matrix
2.1. Host Response to Implantation
2.2. Influence of Material Properties on Cell Adhesion
References
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27.
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869.
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200.
- Kular, J.K.; Basu, S.; Sharma, R.I. The extracellular matrix: Structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J. Tissue Eng. 2014, 5, 112.
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801.
- Halfter, W.; Candiello, J.; Hu, H.; Zhang, P.; Schreiber, E.; Balasubramani, M. Protein composition and biomechanical properties of in vivo-derived basement membranes. Cell Adhes. Migr. 2013, 7, 64–71.
- Pozzi, A.; Yurchenco, P.D.; Iozzo, R.V. The nature and biology of basement membranes. Matrix Biol. 2017, 57–58, 1–11.
- Yurchenco, P.; Patton, B. Developmental and Pathogenic Mechanisms of Basement Membrane Assembly. Curr. Pharm. Des. 2009, 15, 1277–1294.
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616.
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14.
- Keller, K.E.; Kelley, M.J.; Acott, T.S. Extracellular matrix gene alternative splicing by trabecular meshwork cells in response to mechanical stretching. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1164–1172.
- Bornstein, P. Matricellular proteins: An overview. J. Cell Commun. Signal. 2009, 3, 163–165.
- Rosset, E.M.; Bradshaw, A.D. SPARC/osteonectin in mineralized tissue. Matrix Biol. 2016, 52–54, 78–87.
- Bornstein, P. Thrombospondins as matricellular modulators of cell function. J. Clin. Investig. 2001, 107, 929–934.
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327.
- Wilson, S.E.; Torricelli, A.A.M.; Marino, G.K. Corneal epithelial basement membrane: Structure, function and regeneration. Exp. Eye Res. 2020, 194, 108002.
- Kvist, A.J.; Nyström, A.; Hultenby, K.; Sasaki, T.; Talts, J.F.; Aspberg, A. The major basement membrane components localize to the chondrocyte pericellular matrix-A cartilage basement membrane equivalent? Matrix Biol. 2008, 27, 22–33.
- Zhang, Z. Chondrons and the Pericellular Matrix of Chondrocytes. Tissue Eng.-Part B Rev. 2015, 21, 267–277.
- Youn, I.; Choi, J.B.; Cao, L.; Setton, L.A.; Guilak, F. Zonal variations in the three-dimensional morphology of the chondron measured in situ using confocal microscopy. Osteoarthr. Cartil. 2006, 14, 889–897.
- Poole, C.A. Review. Articular cartilage chondrons: Form, function and failure. J. Anat. 1997, 191, 1–13.
- Fraser, S.A.; Crawford, A.; Frazer, A.; Dickinson, S.; Hollander, A.P.; Brook, I.M.; Hatton, P.V. Localization of type VI collagen in tissue-engineered cartilage on polymer scaffolds. Tissue Eng. 2006, 12, 569–577.
- Hynes, R.O.; Naba, A. Overview of the matrisome-An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903.
- Barnes, M. Update on Collagens: What You Need to Know and Consider. Plast. Surg. Nurs. 2019, 39, 112–115.
- Exposito, J.Y.; Valcourt, U.; Cluzel, C.; Lethias, C. The fibrillar collagen family. Int. J. Mol. Sci. 2010, 11, 407–426.
- Bella, J.; Hulmes, D.J.S. Fibrillar collagens. Subcell. Biochem. 2017, 82, 457–490.
- Mienaltowski, M.J.; Birk, D.E. Structure, Physiology, and Biochemistry of Collagens. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 802, pp. 5–29.
- Shaw, L.M.; Olsen, B.R. FACIT collagens: Diverse molecular bridges in extracellular matrices. Trends Biochem. Sci. 1991, 16, 191–194.
- Ricard-Blum, S.; Ruggiero, F. The collagen superfamily: From the extracellular matrix to the cell membrane. Pathol. Biol. 2005, 53, 430–442.
- Ivanova, V.P.; Krivchenko, A.I. Current viewpoint on structure and on evolution of collagens. II. Fibril-associated collagens. J. Evol. Biochem. Physiol. 2014, 50, 273–285.
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958.
- Harsha, L.; Brundha, M.P. Role of collagen in wound healing. Drug Invent. Today 2020, 13, 55–57.
- Heino, J. The collagen family members as cell adhesion proteins. BioEssays 2007, 29, 1001–1010.
- Luckman, S.P.; Rees, E.; Kwan, A.P.L. Partial characterization of cell-type X collagen interactions. Biochem. J. 2003, 372, 485–493.
- Smethurst, P.A.; Onley, D.J.; Jarvis, G.E.; O’Connor, M.N.; Graham Knight, C.; Herr, A.B.; Ouwehand, W.H.; Farndale, R.W. Structural basis for the platelet-collagen interaction: The smallest motif within collagen that recognizes and activates platelet Glycoprotein VI contains two glycine-proline-hydroxyproline triplets. J. Biol. Chem. 2007, 282, 1296–1304.
- Paavola, K.J.; Sidik, H.; Zuchero, J.B.; Eckart, M.; Talbot, W.S. Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Sci. Signal. 2014, 7, ra76.
- Wolf, K.; Alexander, S.; Schacht, V.; Coussens, L.M.; von Andrian, U.H.; van Rheenen, J.; Deryugina, E.; Friedl, P. Collagen-based cell migration models in vitro and in vivo. Semin. Cell Dev. Biol. 2009, 20, 931–941.
- Guido, S.; Tranquillo, R.T. A methodology for the systematic and quantitative study of cell contact guidance in oriented collagen gels. Correlation of fibroblast orientation and gel birefringence. J. Cell Sci. 1993, 105, 317–331.
- Borgne-Rochet, M.L.; Angevin, L.; Bazellières, E.; Ordas, L.; Comunale, F.; Denisov, E.V.; Tashireva, L.A.; Perelmuter, V.M.; Bièche, I.; Vacher, S.; et al. P-cadherin-induced decorin secretion is required for collagen fiber alignment and directional collective cell migration. J. Cell Sci. 2019, 132, 3189.
- Canty, E.G.; Kadler, K.E. Collagen fibril biosynthesis in tendon: A review and recent insights. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2002, 133, 979–985.
- Zhang, G.; Young, B.B.; Birk, D.E. Differential expression of type XII collagen in developing chicken metatarsal tendons. J. Anat. 2003, 202, 411–420.
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–19.
- Parenteau-Bareil, R.; Gauvin, R.; Berthod, F. Collagen-based biomaterials for tissue engineering applications. Materials 2010, 3, 1863–1887.
- Cocciolone, A.J.; Hawes, J.Z.; Staiculescu, M.C.; Johnson, E.O.; Murshed, M.; Wagenseil, J.E. Elastin, arterial mechanics, and cardiovascular disease. Am. J. Physiol.-Hear Circ. Physiol. 2018, 315, H189–H205.
- Mariani, T.J.; Dunsmore, S.E.; Li, Q.; Ye, X.; Pierce, R.A. Regulation of lung fibroblast tropoelastin expression by alveolar epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1998, 274, L47–L57.
- Mecham, R.P.; Madaras, J.; McDonald, J.A.; Ryan, U. Elastin production by cultured calf pulmonary artery endothelial cells. J. Cell. Physiol. 1983, 116, 282–288.
- Kajiya, H.; Tanaka, N.; Inazumi, T.; Seyama, Y.; Tajima, S.; Ishibashi, A. Cultured human keratinocytes express tropoelastin. J. Investig. Dermatol. 1997, 109, 641–644.
- Narayanan, A.S.; Sandberg, L.B.; Ross, R.; Layman, D.L. The smooth muscle cell: III. Elastin synthesis in arterial smooth muscle cell culture. J. Cell Biol. 1976, 68, 411–419.
- Nishizaki, T. PKCϵ Increases Extracellular Elastin and Fibulin-5/DANCE in Dermal Fibroblasts. Cell. Physiol. Biochem. 2018, 46, 291–302.
- Vrhovski, B.; Weiss, A.S. Biochemistry of tropoelastin. Eur. J. Biochem. 1998, 258, 1–18.
- Ozsvar, J.; Yang, C.; Cain, S.A.; Baldock, C.; Tarakanova, A.; Weiss, A.S. Tropoelastin and Elastin Assembly. Front. Bioeng. Biotechnol. 2021, 9, 138.
- Vindin, H.; Mithieux, S.M.; Weiss, A.S. Elastin architecture. Matrix Biol. 2019, 84, 4–16.
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63, 2304–2316.
- Wagenseil, J.E.; Mecham, R.P. New insights into elastic fiber assembly. Birth Defects Res. Part C-Embryo Today Rev. 2007, 81, 229–240.
- Baldwin, A.K.; Simpson, A.; Steer, R.; Cain, S.A.; Kielty, C.M. Elastic fibres in health and disease. Expert Rev. Mol. Med. 2013, 15, 23.
- Thomson, J.; Singh, M.; Eckersley, A.; Cain, S.A.; Sherratt, M.J.; Baldock, C. Fibrillin microfibrils and elastic fibre proteins: Functional interactions and extracellular regulation of growth factors. Semin. Cell Dev. Biol. 2019, 89, 109–117.
- Ritty, T.M.; Ditsios, K.; Starcher, B.C. Distribution of the elastic fiber and associated proteins in flexor tendon reflects function. Anat. Rec. 2002, 268, 430–440.
- Gabriela Espinosa, M.; Catalin Staiculescu, M.; Kim, J.; Marin, E.; Wagenseil, J.E. Elastic Fibers and Large Artery Mechanics in Animal Models of Development and Disease. J. Biomech. Eng. 2018, 140, 0208031.
- Dubick, M.A.; Rucker, R.B.; Cross, C.E.; Last, J.A. Elastin metabolism in rodent lung. BBA-Gen. Subj. 1981, 672, 303–306.
- Davidson, J.M.; Smith, K.; Shibahara, S.; Tolstoshev, P.; Crystal, R.G. Regulation of elastin synthesis in developing sheep nuchal ligament by elastin mRNA levels. J. Biol. Chem. 1982, 257, 747–754.
- Burnett, W.; Finnigan-Bunick, A.; Yoon, K.; Rosenbloom, J. Analysis of elastin gene expression in the developing chick aorta using cloned elastin cDNA. J. Biol. Chem. 1982, 257, 1569–1572.
- Kühl, T.; Mezger, M.; Hausser, I.; Guey, L.T.; Handgretinger, R.; Bruckner-Tuderman, L.; Nyström, A. Collagen VII Half-Life at the Dermal-Epidermal Junction Zone: Implications for Mechanisms and Therapy of Genodermatoses. J. Investig. Dermatol. 2016, 136, 1116–1123.
- Shapiro, S.D.; Endicott, S.K.; Province, M.A.; Pierce, J.A.; Campbell, E.J. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J. Clin. Investig. 1991, 87, 1828–1834.
- Yanagisawa, H.; Wagenseil, J. Elastic fibers and biomechanics of the aorta: Insights from mouse studies. Matrix Biol. 2020, 85–86, 160–172.
- Yanagisawa, H.; Davis, E.C. Unraveling the mechanism of elastic fiber assembly: The roles of short fibulins. Int. J. Biochem. Cell Biol. 2010, 42, 1084–1093.
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55.
- Perrimon, N.; Bernfield, M. Cellular functions of proteoglycans—An overview. Semin. Cell Dev. Biol. 2001, 12, 65–67.
- Hardingham, T.E.; Fosang, A.J. Proteoglycans: Many forms and many functions. FASEB J. 1992, 6, 861–870.
- Rozario, T.; DeSimone, D.W. The Extracellular Matrix In Development and Morphogenesis: A Dynamic View. Dev. Biol. 2010, 341, 126.
- Lindahl, U. A personal voyage through the proteoglycan field. Matrix Biol. 2014, 35, 3–7.
- Vynios, D.H.; Karamanos, N.K.; Tsiganos, C.P. Advances in analysis of glycosaminoglycans: Its application for the assessment of physiological and pathological states of connective tissues. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 781, 21–38.
- Kjellén, L.; Lindahl, U. Proteoglycans: Structures and interactions. Annu. Rev. Biochem. 1991, 60, 443–475.
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482.
- Ma, J.; Cai, H.; Long, X.; Cheng, K.; Xu, X.; Zhang, D.; Li, J. Hyaluronic acid bioinspired polymers for the regulation of cell chondrogenic and osteogenic differentiation. Int. J. Biol. Macromol. 2020, 161, 1011–1020.
- Ghiselli, G. Drug-Mediated Regulation of Glycosaminoglycan Biosynthesis. Med. Res. Rev. 2017, 37, 1051–1094.
- Hardingham, T.E.; Ewins, R.J.F.; Muir, H. Cartilage proteoglycans. Structure and heterogeneity of the protein core and the effects of specific protein modifications on the binding to hyaluronate. Biochem. J. 1976, 157, 127–143.
- Hascall, V.C. Interaction of cartilage proteoglycans with hyaluronic acid. J. Supramol. Cell. Biochem. 1977, 7, 101–120.
- Rosenberg, L.; Hellmann, W.; Kleinschmidt, A.K. Electron microscopic studies of proteoglycan aggregates from bovine articular cartilage. J. Biol. Chem. 1975, 250, 1877–1883.
- Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and function. Biomolecules 2020, 10, 1525.
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5, 18.
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157.
- Dudhia, J. Aggrecan, aging and assembly in articular cartilage. Cell. Mol. Life Sci. 2005, 62, 2241–2256.
- Walimbe, T.; Panitch, A. Proteoglycans in biomedicine: Resurgence of an underexploited class of ECM molecules. Front. Pharmacol. 2020, 10, 1661.
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126, 3799–3804.
- Choi, Y.; Chung, H.; Jung, H.; Couchman, J.R.; Oh, E.S. Syndecans as cell surface receptors: Unique structure equates with functional diversity. Matrix Biol. 2011, 30, 93–99.
- Kolset, S.O.; Tveit, H. Serglycin-Structure and biology. Cell. Mol. Life Sci. 2008, 65, 1073–1085.
- Henningsson, F.; Hergeth, S.; Cortelius, R.; Åbrink, M.; Pejler, G. A role for serglycin proteoglycan in granular retention and processing of mast cell secretory granule components. FEBS J. 2006, 273, 4901–4912.
- Åbrink, M.; Grujic, M.; Pejler, G. Serglycin is essential for maturation of mast cell secretory granule. J. Biol. Chem. 2004, 279, 40897–40905.
- Whitelock, J.M.; Graham, L.D.; Melrose, J.; Murdoch, A.D.; Iozzo, R.V.; Anne Underwood, P. Human perlecan immunopurified from different endothelial cell sources has different adhesive properties for vascular cells. Matrix Biol. 1999, 18, 163–178.
- Sher, I.; Zisman-Rozen, S.; Eliahu, L.; Whitelock, J.M.; Maas-Szabowski, N.; Yamada, Y.; Breitkreutz, D.; Fusenig, N.E.; Arikawa-Hirasawa, E.; Iozzo, R.V.; et al. Targeting perlecan in human keratinocytes reveals novel roles for perlecan in epidermal formation. J. Biol. Chem. 2006, 281, 5178–5187.
- Nugent, M.A.; Nugent, H.M.; Iozzo, R.V.; Sanchack, K.; Edelman, E.R. Perlecan is required to inhibit thrombosis after deep vascular injury and contributes to endothelial cell-mediated inhibition of intimal hyperplasia. Proc. Natl. Acad. Sci. USA 2000, 97, 6722–6727.
- Iozzo, R.V. Basement membrane proteoglycans: From cellar to ceiling. Nat. Rev. Mol. Cell Biol. 2005, 6, 646–656.
- Bezakova, G.; Ruegg, M.A. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 2003, 4, 295–308.
- Zhang, P.; Yang, L.; Li, G.; Jin, Y.; Wu, D.; Wang, Q.M.; Huang, P. Agrin Involvement in Synaptogenesis Induced by Exercise in a Rat Model of Experimental Stroke. Neurorehabil. Neural Repair 2020, 34, 1124–1137.
- Jan, A.T.; Lee, E.J.; Choi, I. Fibromodulin: A regulatory molecule maintaining cellular architecture for normal cellular function. Int. J. Biochem. Cell Biol. 2016, 80, 66–70.
- Ezura, Y.; Chakravarti, S.; Oldberg, A.; Chervoneva, I.; Birk, D.E. Differential expression of lumican and fibromodulin regulate collagen fibrillogenesis in developing mouse tendons. J. Cell Biol. 2000, 151, 779–787.
- Muncie, J.M.; Weaver, V.M. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. In Current Topics in Developmental Biology; Academic Press Inc.: Amsterdam, The Netherlands, 2018; Volume 130, pp. 1–37.
- Davis, B.G. Synthesis of glycoproteins. Chem. Rev. 2002, 102, 579–601.
- Kornfeld, R.; Kornfeld, S. Comparative aspects of glycoprotein structure. Annu. Rev. Biochem. 1976, 45, 217–237.
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785.
- Mecham, R.P. Overview of extracellular matrix. Curr. Protoc. Cell Biol. 2012, 57, 10.
- Dwek, R.A. Glycobiology: Toward understanding the function of sugars. Chem. Rev. 1996, 96, 683–720.
- Preissner, K.T.; Reuning, U. Vitronectin in vascular context: Facets of a multitalented matricellular protein. Semin. Thromb. Hemost. 2011, 37, 408–424.
- Weeterings, C.; Adelmeijer, J.; Myles, T.; De Groot, P.G.; Lisman, T. Glycoprotein Ibα-mediated platelet adhesion and aggregation to immobilized thrombin under conditions of flow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 670–675.
- Brockhausen, I.; Kuhns, W. Role of Glycoproteins of the Immune and Blood Coagulation Systems; Springer: Berlin/Heidelberg, Germany, 1997; pp. 77–84.
- Rudd, P.M.; Elliott, T.; Cresswell, P.; Wilson, I.A.; Dwek, R.A. Glycosylation and the immune system. Science 2001, 291, 2370–2376.
- Jamieson, J.C.; Kaplan, H.A.; Woloski, B.M.R.N.J. Glycoprotein biosynthesis during the acute-phase response to inflammation. Can. J. Biochem. Cell Biol. 1983, 61, 1041–1048.
- Gupta, S.K. Role of zona pellucida glycoproteins during fertilization in humans. J. Reprod. Immunol. 2015, 108, 90–97.
- Timpl, R.; Sasaki, T.; Kostka, G.; Chu, M.L. Fibulins: A versatile family of extracellular matrix proteins. Nat. Rev. Mol. Cell Biol. 2003, 4, 479–489.
- Chiquet-Ehrismann, R.; Tucker, R.P. Tenascins and the importance of adhesion modulation. Cold Spring Harb. Perspect. Biol. 2011, 3, a004960.
- Weisel, J.W. Fibrinogen and fibrin. Adv. Protein Chem. 2005, 70, 247–299.
- Leavesley, D.I.; Kashyap, A.S.; Croll, T.; Sivaramakrishnan, M.; Shokoohmand, A.; Hollier, B.G.; Upton, Z. Vitronectin-Master controller or micromanager? IUBMB Life 2013, 65, 807–818.
- Ganss, B.; Kim, R.H.; Sodek, J. Bone sialoprotein. Crit. Rev. Oral Biol. Med. 1999, 10, 79–98.
- Fatemi, S.H. Reelin glycoprotein: Structure, biology and roles in health and disease. Mol. Psychiatry 2005, 10, 251–257.
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863.
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419.
- Baron, M.; Norman, D.; Willis, A.; Campbell, I.D. Structure of the fibronectin type 1 module. Nature 1990, 345, 642–646.
- Potts, J.R.; Campbell, I.D. Fibronectin structure and assembly. Curr. Opin. Cell Biol. 1994, 6, 648–655.
- McDonald, J.A.; Kelley, D.G.; Broekelmann, T.J. Role of fibronectin in collagen deposition: Fab’ to the gelatin-binding domain of fibronectin inhibits both fibronectin and collagen organization in fibroblast extracellular matrix. J. Cell Biol. 1982, 92, 485–492.
- Tamkun, J.W.; DeSimone, D.W.; Fonda, D.; Patel, R.S.; Buck, C.; Horwitz, A.F.; Hynes, R.O. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 1986, 46, 271–282.
- Mao, Y.; Schwarzbauer, J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005, 24, 389–399.
- Sekiguchi, K.; Hakomori, S. Functional domain structure of fibronectin. Proc. Natl. Acad. Sci. USA 1980, 77, 2661–2665.
- Hymes, J.P.; Klaenhammer, T.R. Stuck in the middle: Fibronectin-binding proteins in gram-positive bacteria. Front. Microbiol. 2016, 7, 1504.
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenes. Tissue Repair 2011, 4, 21.
- Sottile, J.; Hocking, D.C. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol. Biol. Cell 2002, 13, 3546–3559.
- Sakai, T.; Johnson, K.J.; Murozono, M.; Sakai, K.; Magnuson, M.A.; Wieloch, T.; Cronberg, T.; Isshiki, A.; Erickson, H.P.; Fässler, R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat. Med. 2001, 7, 324–330.
- Maurer, L.M.; Ma, W.; Mosher, D.F. Dynamic structure of plasma fibronectin. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 213–227.
- Schwarzbauer, J.E. Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J. Cell Biol. 1991, 113, 1463–1473.
- Schwarzbauer, J.E.; Tamkun, J.W.; Lemischka, I.R.; Hynes, R.O. Three different fibronectin mRNAs arise by alternative splicing within the coding region. Cell 1983, 35, 421–431.
- Engel, J.; Odermatt, E.; Engel, A.; Madri, J.A.; Furthmayr, H.; Rohde, H.; Timpl, R. Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J. Mol. Biol. 1981, 150, 97–120.
- Senyürek, I.; Kempf, W.E.; Klein, G.; Maurer, A.; Kalbacher, H.; Schäfer, L.; Wanke, I.; Christ, C.; Stevanovic, S.; Schaller, M.; et al. Processing of laminin α chains generates peptides involved in wound healing and host defense. J. Innate Immun. 2014, 6, 467–484.
- Aumailley, M.; Bruckner-Tuderman, L.; Carter, W.G.; Deutzmann, R.; Edgar, D.; Ekblom, P.; Engel, J.; Engvall, E.; Hohenester, E.; Jones, J.C.R.; et al. A simplified laminin nomenclature. Matrix Biol. 2005, 24, 326–332.
- Hohenester, E. Structural biology of laminins. Essays Biochem. 2019, 63, 285–295.
- Odenthal, U.; Haehn, S.; Tunggal, P.; Merkl, B.; Schomburg, D.; Frie, C.; Paulsson, M.; Smyth, N. Molecular analysis of laminin N-terminal domains mediating self-interactions. J. Biol. Chem. 2004, 279, 44504–44512.
- Schittny, J.C.; Yurchenco, P.D. Terminal short arm domains of basement membrane laminin are critical for its self-assembly. J. Cell Biol. 1990, 110, 825–832.
- Hussain, S.A.; Carafoli, F.; Hohenester, E. Determinants of laminin polymerization revealed by the structure of the α5 chain amino-terminal region. EMBO Rep. 2011, 12, 276–282.
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adhes. Migr. 2013, 7, 56–63.
- McKee, K.K.; Hohenester, E.; Aleksandrova, M.; Yurchenco, P.D. Organization of the laminin polymer node. Matrix Biol. 2021, 98, 49–63.
- Sasaki, T.; Fässler, R.; Hohenester, E. Laminin: The crux of basement membrane assembly. J. Cell Biol. 2004, 164, 959–963.
- Colognato, H.; Yurchenco, P.D. Form and function: The laminin family of heterotrimers. Dev. Dyn. 2000, 218, 213–234.
- Colognato, H.; Winkelmann, D.A.; Yurchenco, P.D. Laminin polymerization induces a receptor-cytoskeleton network. J. Cell Biol. 1999, 145, 619–631.
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628.
- Costell, M.; Gustafsson, E.; Aszódi, A.; Mörgelin, M.; Bloch, W.; Hunziker, E.; Addicks, K.; Timpl, R.; Fässler, R. Perlecan maintains the integrity of cartilage and some basement membranes. J. Cell Biol. 1999, 147, 1109–1122.
- McKee, K.K.; Aleksandrova, M.; Yurchenco, P.D. Chimeric protein identification of dystrophic, Pierson and other laminin polymerization residues. Matrix Biol. 2018, 67, 32–46.
- Böse, K.; Nischt, R.; Page, A.; Bader, B.L.; Paulsson, M.; Smyth, N. Loss of nidogen-1 and -2 results in syndactyly and changes in limb development. J. Biol. Chem. 2006, 281, 39620–39629.
- Li, S.; Liquari, P.; McKee, K.K.; Harrison, D.; Patel, R.; Lee, S.; Yurchenco, P.D. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J. Cell Biol. 2005, 169, 179–189.
- Mak, K.M.; Mei, R. Basement Membrane Type IV Collagen and Laminin: An Overview of Their Biology and Value as Fibrosis Biomarkers of Liver Disease. Anat. Rec. 2017, 300, 1371–1390.
- Cambi, A.; Chavrier, P. Tissue remodeling by invadosomes. Fac. Rev. 2021, 10, 39.
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254.
- Cornfine, S.; Himmel, M.; Kopp, P.; el Azzouzi, K.; Wiesner, C.; Krüger, M.; Rudel, T.; Linder, S. The kinesin KIF9 and reggie/flotillin proteins regulate matrix degradation by macrophage podosomes. Mol. Biol. Cell 2011, 22, 202.
- Cawston, T.E.; Young, D.A. Proteinases involved in matrix turnover during cartilage and bone breakdown. Cell Tissue Res. 2010, 339, 221–235.
- Oda, K. New families of carboxyl peptidases: Serine-carboxyl peptidases and glutamic peptidases. J. Biochem. 2012, 151, 13–25.
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1.
- Kapoor; Vaidya, S.; Wadhwan, V.; Hitesh; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. Ther. 2016, 12, 28.
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221.
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix metalloproteinases: Inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediators Inflamm. 2013, 2013, 8315.
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 17, 463–516.
- Imai, K.; Hiramatsu, A.; Fukushima, D.; Pierschbacher, M.D.; Okada, Y. Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-β1 release. Biochem. J. 1997, 322, 809–814.
- Giannelli, G.; Falk-Marzillier, J.; Schiraldi, O.; Stetler-Stevenson, W.G.; Quaranta, V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science 1997, 277, 225–228.
- Stetler-Stevenson, W.G. Matrix metalloproteinases in angiogenesis: A moving target for therapeutic intervention. J. Clin. Investig. 1999, 103, 1237–1241.
- Anacker, J.; Segerer, S.E.; Hagemann, C.; Feix, S.; Kapp, M.; Bausch, R.; Kämmerer, U. Human decidua and invasive trophoblasts are rich sources of nearly all human matrix metalloproteinases. Mol. Hum. Reprod. 2011, 17, 637–652.
- Yushchenko, M.; Weber, F.; Mäder, M.; Schöll, U.; Maliszewska, M.; Tumani, H.; Felgenhauer, K.; Beuche, W. Matrix metalloproteinase-9 (MMP-9) in human cerebrospinal fluid (CSF): Elevated levels are primarily related to CSF cell count. J. Neuroimmunol. 2000, 110, 244–251.
- Hartung, H.-P.; Kieseier, B.C. The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J. Neuroimmunol. 2000, 107, 140–147.
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83.
- Blobel, C.P. Metalloprotease-Disintegrins: Links to Cell Adhesion and Cleavage of TNFα and Notch. Cell 1997, 90, 589–592.
- Werb, Z. ECM and Cell Surface Proteolysis: Regulating Cellular Ecology. Cell 1997, 91, 439–442.
- Blobel, C.P. ADAMs: Key components in egfr signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43.
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 929–941.
- Rose-John, S. ADAM17, shedding, TACE as therapeutic targets. Pharmacol. Res. 2013, 71, 19–22.
- Marczynska, J.; Ozga, A.; Wlodarczyk, A.; Majchrzak-Gorecka, M.; Kulig, P.; Banas, M.; Michalczyk-Wetula, D.; Majewski, P.; Hutloff, A.; Schwarz, J.; et al. The Role of Metalloproteinase ADAM17 in Regulating ICOS Ligand–Mediated Humoral Immune Responses. J. Immunol. 2014, 193, 2753–2763.
- Hsia, H.E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell. Mol. Life Sci. 2019, 76, 3055–3081.
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 2059–2070.
- Van Goor, H.; Melenhorst, W.B.W.H.; Turner, A.J.; Holgate, S.T. Adamalysins in biology and disease. J. Pathol. 2009, 219, 277–286.
- Boudreau, N.J.; Jones, P.L. Extracellular matrix and integrin signalling: The shape of things to come. Biochem. J. 1999, 339, 481–488.
- Fan, D.; Creemers, E.E.; Kassiri, Z. Matrix as an interstitial transport system. Circ. Res. 2014, 114, 889–902.
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162.
- Flaumenhaft, R.; Rifkin, D.B. Extracellular matrix regulation of growth factor and protease activity. Curr. Opin. Cell Biol. 1991, 3, 817–823.
- O’Callaghan, P.; Zhang, X.; Li, J.P. Heparan Sulfate Proteoglycans as Relays of Neuroinflammation. J. Histochem. Cytochem. 2018, 66, 305–319.
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. In International Review of Cell and Molecular Biology; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 325, pp. 215–273. ISBN 9780128048061.
- Billings, P.C.; Pacifici, M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: Mechanisms and mysteries. Connect. Tissue Res. 2015, 56, 272–280.
- Matsuo, I.; Kimura-Yoshida, C. Extracellular distribution of diffusible growth factors controlled by heparan sulfate proteoglycans during mammalian embryogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130545.
- Xu, Y.H.; Zhu, Y.; Zhu, Y.Y.; Wei, H.; Zhang, N.N.; Qin, J.S.; Zhu, X.L.; Yu, M.; Li, Y.F. Abnormalities in FGF family members and their roles in modulating depression-related molecules. Eur. J. Neurosci. 2021, 53, 140–150.
- Malkowski, A.; Sobolewski, K.; Jaworski, S.; Bankowski, E. FGF binding by extracellular matrix components of Wharton’s jelly. Acta Biochim. Pol. 2007, 54, 357–363.
- Loo, B.M.; Kreuger, J.; Jalkanen, M.; Lindahl, U.; Salmivirta, M. Binding of Heparin/Heparan Sulfate to Fibroblast Growth Factor Receptor 4. J. Biol. Chem. 2001, 276, 16868–16876.
- Vogler, E.A. Structure and reactivity of water at biomaterial surfaces. Adv. Colloid Interface Sci. 1998, 74, 69–117.
- Kasemo, B.; Gold, J. Implant surfaces and interface processes. Adv. Dent. Res. 1999, 13, 8–20.
- Rabe, M.; Verdes, D.; Seeger, S. Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106.
- Andrade, J.D.; Hlady, V.; Wei, A.P. Adsorption of complex proteins at interfaces. Pure Appl. Chem. 1992, 64, 1777–1781.
- Felgueiras, H.P.; Antunes, J.C.; Martins, M.C.L.; Barbosa, M.A. Fundamentals of protein and cell interactions in biomaterials. In Peptides and Proteins as Biomaterials for Tissue Regeneration and Repair; Barbosa, M.A., Martins, M.C., Eds.; Woodhead Publishing: Thurston, UK, 2018; pp. 1–27. ISBN 9780081008522.
- Norde, W.; Giacomelli, C.E. Conformational changes in proteins at interfaces: From solution to the interface, and back. Macromol. Symp. 1999, 145, 125–136.
- Norde, W.; Giacomelli, C.E. BSA structural changes during homomolecular exchange between the adsorbed and the dissolved states. J. Biotechnol. 2000, 79, 259–268.
- Lundstrom, I. Models of Protein Adsorption on Solid Surfaces. Prog. Colloid Polym. Sci. 1985, 70, 76–82.
- Mariani, E.; Lisignoli, G.; Borzì, R.M.; Pulsatelli, L. Biomaterials: Foreign bodies or tuners for the immune response? Int. J. Mol. Sci. 2019, 20, 636.
- Ode Boni, B.O.; Lamboni, L.; Souho, T.; Gauthier, M.; Yang, G. Immunomodulation and cellular response to biomaterials: The overriding role of neutrophils in healing. Mater. Horizons 2019, 6, 1122–1137.
- Jhunjhunwala, S.; Aresta-DaSilva, S.; Tang, K.; Alvarez, D.; Webber, M.J.; Tang, B.C.; Lavin, D.M.; Veiseh, O.; Doloff, J.C.; Bose, S.; et al. Neutrophil responses to sterile implant materials. PLoS ONE 2015, 10, e0137550.
- Desalegn, G.; Pabst, O. Inflammation triggers immediate rather than progressive changes in monocyte differentiation in the small intestine. Nat. Commun. 2019, 10, 1–14.
- Mesure, L.; de Visscher, G.; Vranken, I.; Lebacq, A.; Flameng, W. Gene expression study of monocytes/macrophages during early foreign body reaction and identification of potential precursors of myofibroblasts. PLoS ONE 2010, 5, e12949.
- Christenson, E.M.; Dadsetan, M.; Anderson, J.M.; Hiltner, A. Biostability and macrophage-mediated foreign body reaction of silicone-modified polyurethanes. J. Biomed. Mater. Res. Part A 2005, 74, 141–155.
- Sheikh, Z.; Brooks, P.J.; Barzilay, O.; Fine, N.; Glogauer, M. Macrophages, foreign body giant cells and their response to implantable biomaterials. Materials 2015, 8, 5671–5701.
- Ward, W.K.; Slobodzian, E.P.; Tiekotter, K.L.; Wood, M.D. The effect of microgeometry, implant thickness and polyurethane chemistry on the foreign body response to subcutaneous implants. Biomaterials 2002, 23, 4185–4192.
- Hulbert, S.F.; Morrison, S.J.; Klawitter, J.J. Tissue reaction to three ceramics of porous and non-porous structures. J. Biomed. Mater. Res. 1972, 6, 347–374.
- Zdolsek, J.; Eaton, J.W.; Tang, L. Histamine release and fibrinogen adsorption mediate acute inflammatory responses to biomaterial implants in humans. J. Transl. Med. 2007, 5, 31.
- Theoharides, T.C.; Tsilioni, I.; Conti, P. Mast cells may regulate the anti-inflammatory activity of IL-37. Int. J. Mol. Sci. 2019, 20, 3701.
- Thevenot, P.T.; Nair, A.M.; Shen, J.; Lotfi, P.; Ko, C.Y.; Tang, L. The effect of incorporation of SDF-1α into PLGA scaffolds on stem cell recruitment and the inflammatory response. Biomaterials 2010, 31, 3997–4008.
- Zachman, A.L.; Crowder, S.W.; Ortiz, O.; Zienkiewicz, K.J.; Bronikowski, C.M.; Yu, S.S.; Giorgio, T.D.; Guelcher, S.A.; Kohn, J.; Sung, H.J. Pro-angiogenic and anti-inflammatory regulation by functional peptides loaded in polymeric implants for soft tissue regeneration. Tissue Eng.-Part A 2013, 19, 437–447.
- Hosoyama, K.; Ahumada, M.; Goel, K.; Ruel, M.; Suuronen, E.J.; Alarcon, E.I. Electroconductive materials as biomimetic platforms for tissue regeneration. Biotechnol. Adv. 2019, 37, 444–458.
- Mason, T.O.; Shimanovich, U. Fibrous Protein Self-Assembly in Biomimetic Materials. Adv. Mater. 2018, 30, 1706462.
- Navarro, M.; Michiardi, A.; Castaño, O.; Planell, J.A. Biomaterials in orthopaedics. J. R. Soc. Interface 2008, 5, 1137–1158.
- Xu, L.C.; Siedlecki, C.A. Effects of surface wettability and contact time on protein adhesion to biomaterial surfaces. Biomaterials 2007, 28, 3273–3283.
- Parhi, P.; Golas, A.; Barnthip, N.; Noh, H.; Vogler, E.A. Volumetric interpretation of protein adsorption: Capacity scaling with adsorbate molecular weight and adsorbent surface energy. Biomaterials 2009, 30, 6814–6824.
- Thevenot, P.; Hu, W.; Tang, L. Surface chemistry influences implant biocompatibility. Curr. Top. Med. Chem. 2008, 8, 270–280.
- Roach, P.; Farrar, D.; Perry, C.C. Interpretation of protein adsorption: Surface-induced conformational changes. J. Am. Chem. Soc. 2005, 127, 8168–8173.
- Roach, P.; Parker, T.; Gadegaard, N.; Alexander, M.R. Surface strategies for control of neuronal cell adhesion: A review. Surf. Sci. Rep. 2010, 65, 145–173.
- Chen, S.; Guo, Y.; Liu, R.; Wu, S.; Fang, J.; Huang, B.; Li, Z.; Chen, Z.; Chen, Z. Tuning surface properties of bone biomaterials to manipulate osteoblastic cell adhesion and the signaling pathways for the enhancement of early osseointegration. Colloids Surfaces B Biointerfaces 2018, 164, 58–69.
- Zelzer, M.; Albutt, D.; Alexander, M.R.; Russell, N.A. The Role of Albumin and Fibronectin in the Adhesion of Fibroblasts to Plasma Polymer Surfaces. Plasma Process. Polym. 2012, 9, 149–156.
- Guo, B.; Anzai, J.I.; Osa, T. Adsorption behavior of serum albumin on electrode surfaces and the effects of electrode potential. Chem. Pharm. Bull. 1996, 44, 800–803.
- Faucheux, N.; Schweiss, R.; Lützow, K.; Werner, C.; Groth, T. Self-assembled monolayers with different terminating groups as model substrates for cell adhesion studies. Biomaterials 2004, 25, 2721–2730.
- Keselowsky, B.G.; Collard, D.M.; García, A.J. Integrin binding specificity regulates biomaterial surface chemistry effects on cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 5953–5957.
- Lan, M.A.; Gersbach, C.A.; Michael, K.E.; Keselowsky, B.G.; García, A.J. Myoblast proliferation and differentiation on fibronectin-coated self assembled monolayers presenting different surface chemistries. Biomaterials 2005, 26, 4523–4531.
- McClary, K.B.; Ugarova, T.; Grainger, D.W. Modulating fibroblast adhesion, spreading, and proliferation using self- assembled monolayer films of alkylthiolates on gold. J. Biomed. Mater. Res. 2000, 50, 428–439.
- Aiyelabegan, H.T.; Sadroddiny, E. Fundamentals of protein and cell interactions in biomaterials. Biomed. Pharmacother. 2017, 88, 956–970.
- Ruoslahti, E.; Pierschbacher, M.D. Arg-Gly-Asp: A versatile cell recognition signal. Cell 1986, 44, 517–518.
- Karimi, F.; O’Connor, A.J.; Qiao, G.G.; Heath, D.E. Integrin Clustering Matters: A Review of Biomaterials Functionalized with Multivalent Integrin-Binding Ligands to Improve Cell Adhesion, Migration, Differentiation, Angiogenesis, and Biomedical Device Integration. Adv. Healthc. Mater. 2018, 7, 1701324.
- Reyes, C.D.; García, A.J. Engineering integrin-specific surfaces with a triple-helical collagen-mimetic peptide. J. Biomed. Mater. Res.-Part A 2003, 65, 511–523.
- Nomizu, M.; Weeks, B.S.; Weston, C.A.; Kim, W.H.; Kleinman, H.K.; Yamada, Y. Structure-activity study of a laminin α1 chain active peptide segment Ile-Lys-Val-Ala-Val (IKVAV). FEBS Lett. 1995, 365, 227–231.
- Yin, Y.; Wang, W.; Shao, Q.; Li, B.; Yu, D.; Zhou, X.; Parajuli, J.; Xu, H.; Qiu, T.; Yetisen, A.K.; et al. Pentapeptide IKVAV-engineered hydrogels for neural stem cell attachment. Biomater. Sci. 2021, 9, 2887–2892.
- Boateng, S.Y.; Lateef, S.S.; Mosley, W.; Hartman, T.J.; Hanley, L.; Russell, B. RGD and YIGSR synthetic peptides facilitate cellular adhesion identical to that of laminin and fibronectin but alter the physiology of neonatal cardiac myocytes. Am. J. Physiol.-Cell Physiol. 2005, 288, 30–38.
- Aota, S.I.; Nomizu, M.; Yamada, K.M. The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J. Biol. Chem. 1994, 269, 24756–24761.
- Cutler, S.M.; García, A.J. Engineering cell adhesive surfaces that direct integrin α5β1 binding using a recombinant fragment of fibronectin. Biomaterials 2003, 24, 1759–1770.
- Lebaron, R.G.; Athanasiou, K.A. Extracellular matrix cell adhesion peptides: Functional applications in orthopedic materials. Tissue Eng. 2000, 6, 85–103.
- Song, W.; Mano, J.F. Interactions between cells or proteins and surfaces exhibiting extreme wettabilities. Soft Matter 2013, 9, 2985–2999.
- Siebers, M.C.; Ter Brugge, P.J.; Walboomers, X.F.; Jansen, J.A. Integrins as linker proteins between osteoblasts and bone replacing materials. A critical review. Biomaterials 2005, 26, 137–146.
- Ponche, A.; Bigerelle, M.; Anselme, K. Relative influence of surface topography and surface chemistry on cell response to bone implant materials. Part 1: Physico-chemical effects. J. Eng. Med. 2010, 224, 1471–1486.
- Ayala, P.; Desai, T.A. Integrin α3 blockade enhances microtopographical down-regulation of α-smooth muscle actin: Role of microtopography in ECM regulation. Integr. Biol. 2011, 3, 733–741.
- Jäger, M.; Zilkens, C.; Zanger, K.; Krauspe, R. Significance of nano- and microtopography for cell-surface interactions in orthopaedic implants. J. Biomed. Biotechnol. 2007, 2007, 69036.
- Hulander, M.; Lundgren, A.; Berglin, M.; Ohrlander, M.; Lausmaa, J.; Elwing, H. Immune complement activation is attenuated by surface nanotopography. Int. J. Nanomed. 2011, 6, 2653–2666.
- Draghi, L.; Cigada, A. Nanostructured surfaces for biomedical applications. Part I: Nanotopography. J. Appl. Biomater. Biomech. 2007, 5, 61–69.
- Folch, A.; Toner, M. Microengineering of cellular interactions. Annu. Rev. Biomed. Eng. 2000, 2, 227–256.
- Poellmann, M.J.; Harrell, P.A.; King, W.P.; Johnson, A.J.W. Geometric microenvironment directs cell morphology on topographically patterned hydrogel substrates. Acta Biomater. 2010, 6, 3514–3523.
- Solanki, A.; Shah, S.; Memoli, K.A.; Park, S.Y.; Hong, S.; Lee, K.B. Controlling differentiation of neural stem cells using extracellular matrix protein patterns. Small 2010, 6, 2509–2513.
- Carthew, J.; Abdelmaksoud, H.H.; Hodgson-Garms, M.; Aslanoglou, S.; Ghavamian, S.; Elnathan, R.; Spatz, J.P.; Brugger, J.; Thissen, H.; Voelcker, N.H.; et al. Precision Surface Microtopography Regulates Cell Fate via Changes to Actomyosin Contractility and Nuclear Architecture. Adv. Sci. 2021, 8, 2003186.
- Anselme, K.; Ploux, L.; Ponche, A. Cell/material interfaces: Influence of surface chemistry and surface topography on cell adhesion. J. Adhes. Sci. Technol. 2010, 24, 831–852.
- Majhy, B.; Priyadarshini, P.; Sen, A.K. Effect of surface energy and roughness on cell adhesion and growth-facile surface modification for enhanced cell culture. RSC Adv. 2021, 11, 15467–15476.
- Hallab, N.J.; Bundy, K.J.; O’Connor, K.; Moses, R.L.; Jacobs, J.J. Evaluation of metallic and polymeric biomaterial surface energy and surface roughness characteristics for directed cell adhesion. Tissue Eng. 2001, 7, 55–70.
- Hou, Y.; Xie, W.; Yu, L.; Camacho, L.C.; Nie, C.; Zhang, M.; Haag, R.; Wei, Q. Surface Roughness Gradients Reveal Topography-Specific Mechanosensitive Responses in Human Mesenchymal Stem Cells. Small 2020, 16, 1905422.