Neutrophils and their products are increasingly recognized to have a key influence on cancer progression and response to therapy. Their involvement has been shown in nearly every aspect of cancer pathophysiology with growing evidence now supporting their role in resistance to a variety of cancer therapies. Recently, the role of neutrophils in cancer progression and therapy resistance has been further complicated with the discovery of neutrophil extracellular traps (NETs). NETs are web-like structures of chromatin decorated with a variety of microbicidal proteins. They are released by neutrophils in a process called NETosis. NET-dependent mechanisms of cancer pathology are beginning to be appreciated, particularly with respect to tumor response to chemo-, immuno-, and radiation therapy.
- neutrophil extracellular traps
- cancer therapy
- inflammation
1. NETs in Chemotherapy Resistance
2. NETs in Immunotherapy Resistance
3. NETs in Radiation Therapy Resistance
4. NET Components in Cancer Therapy Resistance
4.1. Neutrophil Elastase
4.2. Matrix Metalloproteinase 9
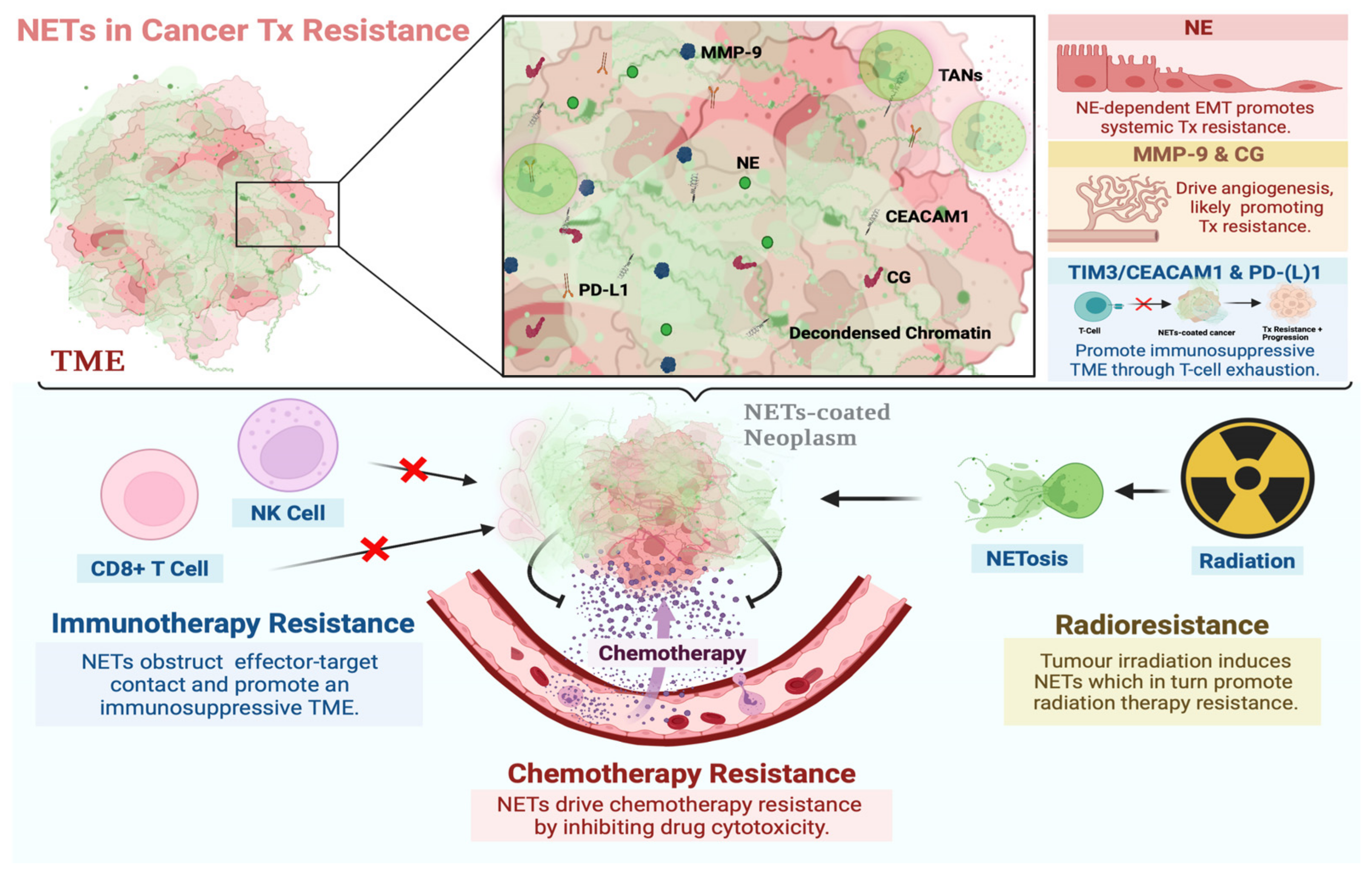
Figure 1. Involvement of NETs in resistance to systemic and local cancer therapies. NETs are DNA webs extruded by neutrophils and associated with NE, MMP-9, CG, CEACAM1, and PD-L1 proteins, among others. These proteins are known to drive mechanisms associated with neoplastic resistance to systemic and local therapies. NETs may promote a microenvironment that favors the development of such a phenotype by sequestering tumor cells, bringing them into contact with these proteins, and driving multiple mechanisms of resistance concurrently. The functional role of NETs in resistance to chemo-, immuno-, and radiation therapy has been reported [61][65][66][67][68]. Proposed mechanisms thereof include NETs coating neoplasms and preventing contact with cytotoxic immune cells, T-cell exhaustion through CEACAM1/TIM-3 interactions and the PD-1/PD-L1 axis, drug detoxification, angiogenesis through CG and MMP-9 activity, and NE-dependent EMT, among others. Created with BioRender.com (accessed on 28 January 2022).
4.3. Cathepsin G
4.4. Carcinoembryonic Antigen Cell Adhesion Molecule 1
4.5. PD-(L)1
References
- Lin, C.; Herlihy, S.E.; Li, M.; Deng, H.; Bernabei, L.; Gabrilovich, D.I.; Vogl, D.T.; Nefedova, Y. NETs promote tumor resistance to anthracyclines. In Proceedings of the American Association for Cancer Research Annual Meeting, Altanta, GA, USA, 29 March–3 April 2019; p. 2103.
- Teijeira, Á.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e858.
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17–induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e2019035.
- Shinde-Jadhav, S.; Mansure, J.J.; Rayes, R.F.; Marcq, G.; Ayoub, M.; Skowronski, R.; Kool, R.; Bourdeau, F.; Brimo, F.; Spicer, J.; et al. Role of neutrophil extracellular traps in radiation resistance of invasive bladder cancer. Nat. Commun. 2021, 12, 2776.
- Kaltenmeier, C.; Yazdani, H.O.; Morder, K.; Geller, D.A.; Simmons, R.L.; Tohme, S. Neutrophil Extracellular Traps Promote T Cell Exhaustion in the Tumor Microenvironment. Front. Immunol. 2021, 12, 785222.
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092.
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031.
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546.
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290.
- Paz-Ares, L.; Ciuleanu, T.E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211.
- Provencio, M.; Nadal, E.; Insa, A.; García-Campelo, M.R.; Casal-Rubio, J.; Dómine, M.; Majem, M.; Rodríguez-Abreu, D.; Martínez-Martí, A.; De Castro Carpeño, J.; et al. Neoadjuvant chemotherapy and nivolumab in resectable non-small-cell lung cancer (NADIM): An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1413–1422.
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833.
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532.
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235.
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551.
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742.
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccin. Immunother. 2019, 15, 1111–1122.
- Zhang, Y.; Guoqiang, L.; Sun, M.; Lu, X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol. Med. 2020, 17, 32–43.
- Teijeira, Á.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e858.
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17–induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e2019035.
- Sasaki, A.; Nakamura, Y.; Mishima, S.; Kawazoe, A.; Kuboki, Y.; Bando, H.; Kojima, T.; Doi, T.; Ohtsu, A.; Yoshino, T.; et al. Predictive factors for hyperprogressive disease during nivolumab as anti-PD1 treatment in patients with advanced gastric cancer. Gastric Cancer 2019, 22, 793–802.
- Wisdom, A.J.; Hong, C.S.; Lin, A.J.; Xiang, Y.; Cooper, D.E.; Zhang, J.; Xu, E.S.; Kuo, H.C.; Mowery, Y.M.; Carpenter, D.J.; et al. Neutrophils promote tumor resistance to radiation therapy. Proc. Natl. Acad. Sci. USA 2019, 116, 18584–18589.
- Moding, E.J.; Kastan, M.B.; Kirsch, D.G. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat. Rev. Drug Discov. 2013, 12, 526–542.
- Kim, B.M.; Hong, Y.; Lee, S.; Liu, P.; Lim, J.H.; Lee, Y.H.; Lee, T.H.; Chang, K.T.; Hong, Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26880–26913.
- Shinde-Jadhav, S.; Mansure, J.J.; Rayes, R.F.; Marcq, G.; Ayoub, M.; Skowronski, R.; Kool, R.; Bourdeau, F.; Brimo, F.; Spicer, J.; et al. Role of neutrophil extracellular traps in radiation resistance of invasive bladder cancer. Nat. Commun. 2021, 12, 2776.
- Cools-Lartigue, J.; Spicer, J.; Najmeh, S.; Ferri, L. Neutrophil extracellular traps in cancer progression. Cell. Mol. Life Sci. 2014, 71, 4179–4194.
- Huang, H.; Zhang, H.; Onuma, A.E.; Tsung, A. Neutrophil Elastase and Neutrophil Extracellular Traps in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1263, 13–23.
- Lerman, I.; Hammes, S.R. Neutrophil elastase in the tumor microenvironment. Steroids 2018, 133, 96–101.
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147.
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691.
- Martins-Cardoso, K.; Almeida, V.H.; Bagri, K.M.; Rossi, M.I.D.; Mermelstein, C.S.; König, S.; Monteiro, R.Q. Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial-Mesenchymal Transition. Cancers 2020, 12, 1542.
- Zhu, T.; Zou, X.; Yang, C.; Li, L.; Wang, B.; Li, R.; Li, H.; Xu, Z.; Huang, D.; Wu, Q. Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelial-mesenchymal transition. Int. J. Mol. Med. 2021, 48, 127.
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374.
- Yeldag, G.; Rice, A.; Del Río Hernández, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471.
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373.
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84.
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773.
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823.
- Peng, Z.; Wang, C.X.; Fang, E.H.; Wang, G.B.; Tong, Q. Role of epithelial-mesenchymal transition in gastric cancer initiation and progression. World J. Gastroenterol. 2014, 20, 5403–5410.
- Deng, J.; Wang, L.; Chen, H.; Hao, J.; Ni, J.; Chang, L.; Duan, W.; Graham, P.; Li, Y. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget 2016, 7, 55771–55788.
- Dong, J.; Wang, R.; Ren, G.; Li, X.; Wang, J.; Sun, Y.; Liang, J.; Nie, Y.; Wu, K.; Feng, B.; et al. HMGA2-FOXL2 Axis Regulates Metastases and Epithelial-to-Mesenchymal Transition of Chemoresistant Gastric Cancer. Clin. Cancer Res. 2017, 23, 3461–3473.
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476.
- Gu, Y.; Zhang, B.; Gu, G.; Yang, X.; Qian, Z. Metformin Increases the Chemosensitivity of Pancreatic Cancer Cells to Gemcitabine by Reversing EMT Through Regulation DNA Methylation of miR-663. Onco Targets Ther. 2020, 13, 10417–10429.
- Yu, J.; Zhang, W.; Gao, F.; Liu, Y.X.; Chen, Z.Y.; Cheng, L.Y.; Xie, S.F.; Zheng, S.S. FBW7 increases chemosensitivity in hepatocellular carcinoma cells through suppression of epithelial-mesenchymal transition. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 184–191.
- Zeng, D.; Liang, Y.K.; Xiao, Y.S.; Wei, X.L.; Lin, H.Y.; Wu, Y.; Bai, J.W.; Chen, M.; Zhang, G.J. Inhibition of Notch1 reverses EMT and chemoresistance to cisplatin via direct downregulation of MCAM in triple-negative breast cancer cells. Int. J. Cancer 2020, 147, 490–504.
- Deryugina, E.; Carré, A.; Ardi, V.; Muramatsu, T.; Schmidt, J.; Pham, C.; Quigley, J.P. Neutrophil Elastase Facilitates Tumor Cell Intravasation and Early Metastatic Events. iScience 2020, 23, 101799.
- Gaida, M.M.; Steffen, T.G.; Günther, F.; Tschaharganeh, D.F.; Felix, K.; Bergmann, F.; Schirmacher, P.; Hänsch, G.M. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur. J. Immunol. 2012, 42, 3369–3380.
- Grosse-Steffen, T.; Giese, T.; Giese, N.; Longerich, T.; Schirmacher, P.; Hänsch, G.M.; Gaida, M.M. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: The role of neutrophils and neutrophil-derived elastase. Clin. Dev. Immunol. 2012, 2012, 720768.
- Mayer, C.; Darb-Esfahani, S.; Meyer, A.S.; Hübner, K.; Rom, J.; Sohn, C.; Braicu, I.; Sehouli, J.; Hänsch, G.M.; Gaida, M.M. Neutrophil Granulocytes in Ovarian Cancer—Induction of Epithelial-To-Mesenchymal-Transition and Tumor Cell Migration. J. Cancer 2016, 7, 546–554.
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739.
- Deryugina, E.I.; Zajac, E.; Juncker-Jensen, A.; Kupriyanova, T.A.; Welter, L.; Quigley, J.P. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia 2014, 16, 771–788.
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474.
- Hawinkels, L.J.A.C.; Zuidwijk, K.; Verspaget, H.W.; de Jonge-Muller, E.S.M.; Duijn, W.V.; Ferreira, V.; Fontijn, R.D.; David, G.; Hommes, D.W.; Lamers, C.B.H.W.; et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur. J. Cancer 2008, 44, 1904–1913.
- Mittal, V.; El Rayes, T.; Narula, N.; McGraw, T.E.; Altorki, N.K.; Barcellos-Hoff, M.H. The Microenvironment of Lung Cancer and Therapeutic Implications. Adv. Exp. Med. Biol. 2016, 890, 75–110.
- Nalla, A.K.; Gorantla, B.; Gondi, C.S.; Lakka, S.S.; Rao, J.S. Targeting MMP-9, uPAR, and cathepsin B inhibits invasion, migration and activates apoptosis in prostate cancer cells. Cancer Gene Ther. 2010, 17, 599–613.
- Gao, H.; Lan, X.; Li, S.; Xue, Y. Relationships of MMP-9, E-cadherin, and VEGF expression with clinicopathological features and response to chemosensitivity in gastric cancer. Tumour. Biol. 2017, 39, 1010428317698368.
- Yang, Y.; Ma, L.; Xu, Y.; Liu, Y.; Li, W.; Cai, J.; Zhang, Y. Enalapril overcomes chemoresistance and potentiates antitumor efficacy of 5-FU in colorectal cancer by suppressing proliferation, angiogenesis, and NF-κB/STAT3-regulated proteins. Cell Death Dis. 2020, 11, 477.
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529.
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426.
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219.
- Wang, S.; Li, Y.; Xing, C.; Ding, C.; Zhang, H.; Chen, L.; You, L.; Dai, M.; Zhao, Y. Tumor microenvironment in chemoresistance, metastasis and immunotherapy of pancreatic cancer. Am. J. Cancer Res. 2020, 10, 1937–1953.
- Bui, T.M.; Yalom, L.K.; Sumagin, R. Tumor-associated neutrophils: Orchestrating cancer pathobiology and therapeutic resistance. Expert Opin. Ther. Targets 2021, 25, 573–583.
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; de Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879.
- Wilson, T.J.; Nannuru, K.C.; Futakuchi, M.; Singh, R.K. Cathepsin G-mediated enhanced TGF-beta signaling promotes angiogenesis via upregulation of VEGF and MCP-1. Cancer Lett. 2010, 288, 162–169.
- Brown, Y.; Hua, S.; Tanwar, P.S. Extracellular matrix-mediated regulation of cancer stem cells and chemoresistance. Int. J. Biochem. Cell Biol. 2019, 109, 90–104.
- Guan, X.; Lu, Y.; Zhu, H.; Yu, S.; Zhao, W.; Chi, X.; Xie, C.; Yin, Z. The Crosstalk Between Cancer Cells and Neutrophils Enhances Hepatocellular Carcinoma Metastasis via Neutrophil Extracellular Traps-Associated Cathepsin G Component: A Potential Therapeutic Target. J. Hepatocell. Carcinoma 2021, 8, 451–465.
- Khan, M.; Carmona, S.; Sukhumalchandra, P.; Roszik, J.; Philips, A.; Perakis, A.A.; Kerros, C.; Zhang, M.; Qiao, N.; John, L.S.S.; et al. Cathepsin G Is Expressed by Acute Lymphoblastic Leukemia and Is a Potential Immunotherapeutic Target. Front. Immunol. 2018, 8, 1975.
- Rayes, R.F.; Vourtzoumis, P.; Bou Rjeily, M.; Seth, R.; Bourdeau, F.; Giannias, B.; Berube, J.; Huang, Y.H.; Rousseau, S.; Camilleri-Broet, S.; et al. Neutrophil Extracellular Trap-Associated CEACAM1 as a Putative Therapeutic Target to Prevent Metastatic Progression of Colon Carcinoma. J. Immunol. 2020, 204, 2285–2294.
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671.
- Dankner, M.; Gray-Owen, S.D.; Huang, Y.H.; Blumberg, R.S.; Beauchemin, N. CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology 2017, 6, e1328336.
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390.
- Ortenberg, R.; Sapoznik, S.; Zippel, D.; Shapira-Frommer, R.; Itzhaki, O.; Kubi, A.; Zikich, D.; Besser, M.J.; Schachter, J.; Markel, G. Serum CEACAM1 Elevation Correlates with Melanoma Progression and Failure to Respond to Adoptive Cell Transfer Immunotherapy. J. Immunol. Res. 2015, 2015, 902137.
- Yang, F.; Zeng, Z.; Li, J.; Ren, X.; Wei, F. TIM-3 and CEACAM1 are Prognostic Factors in Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2021, 8, 619765.
- Zhang, Y.; Cai, P.; Li, L.; Shi, L.; Chang, P.; Liang, T.; Yang, Q.; Liu, Y.; Wang, L.; Hu, L. Co-expression of TIM-3 and CEACAM1 promotes T cell exhaustion in colorectal cancer patients. Int. Immunopharmacol. 2017, 43, 210–218.
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499.
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723.
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2021, 11, 622509.
- Kaltenmeier, C.; Yazdani, H.O.; Morder, K.; Geller, D.A.; Simmons, R.L.; Tohme, S. Neutrophil Extracellular Traps Promote T Cell Exhaustion in the Tumor Microenvironment. Front. Immunol. 2021, 12, 785222.