Von Hippel-Lindau disease (VHL disease or VHL syndrome) is a familial multisystem neoplastic disorder, stemming from germline disease-associated variants of the VHL tumor suppressor gene. VHL protein is involved in oxygen sensing and adaptive response to hypoxia through the EPO-VHL-HIF signaling axis. In recent years, numerous HIF-independent pathways of VHL have been identified, expanding the role of VHL throughout several cellular processes. In addition to VHL syndrome-associated tumors, VHL variations have also been associated with the development of eythrocytosis. Research indicated that there is a distinction between erythrocytosis-causing VHL variations and VHL variations causing VHL disease with tumor development. Therefore, elucidating the molecular background of the pathogenic effects of VHL variants could help determine the best approach to VHL disease management.
- VHL
- VHL disease
- Chuvash polycythemia
- genetic variation
- erythrocytosis
- pheochromocytoma
- renal cell carcinoma
- retinal hemangioblastoma
- hemangioblastoma
1. Introduction
2. VHL Canonical and Non-Canonical Functions
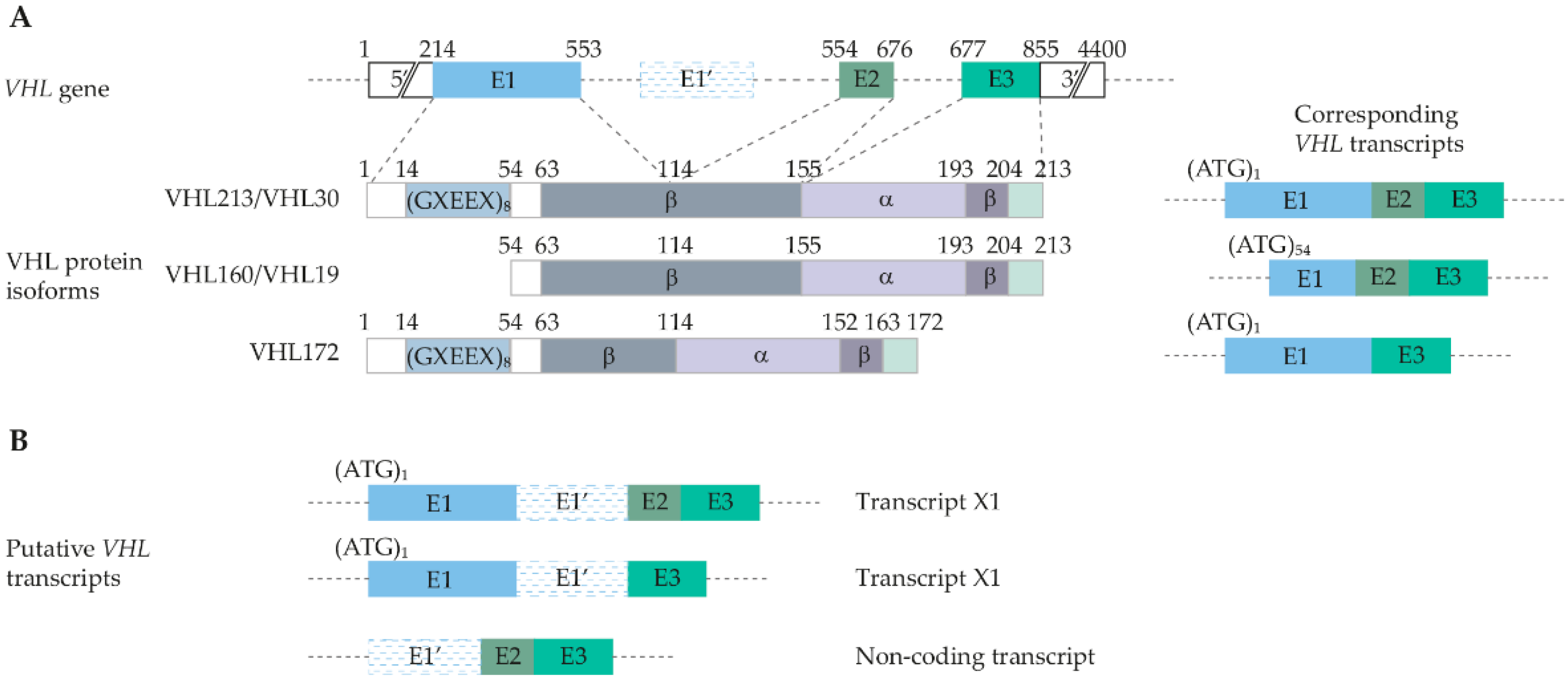
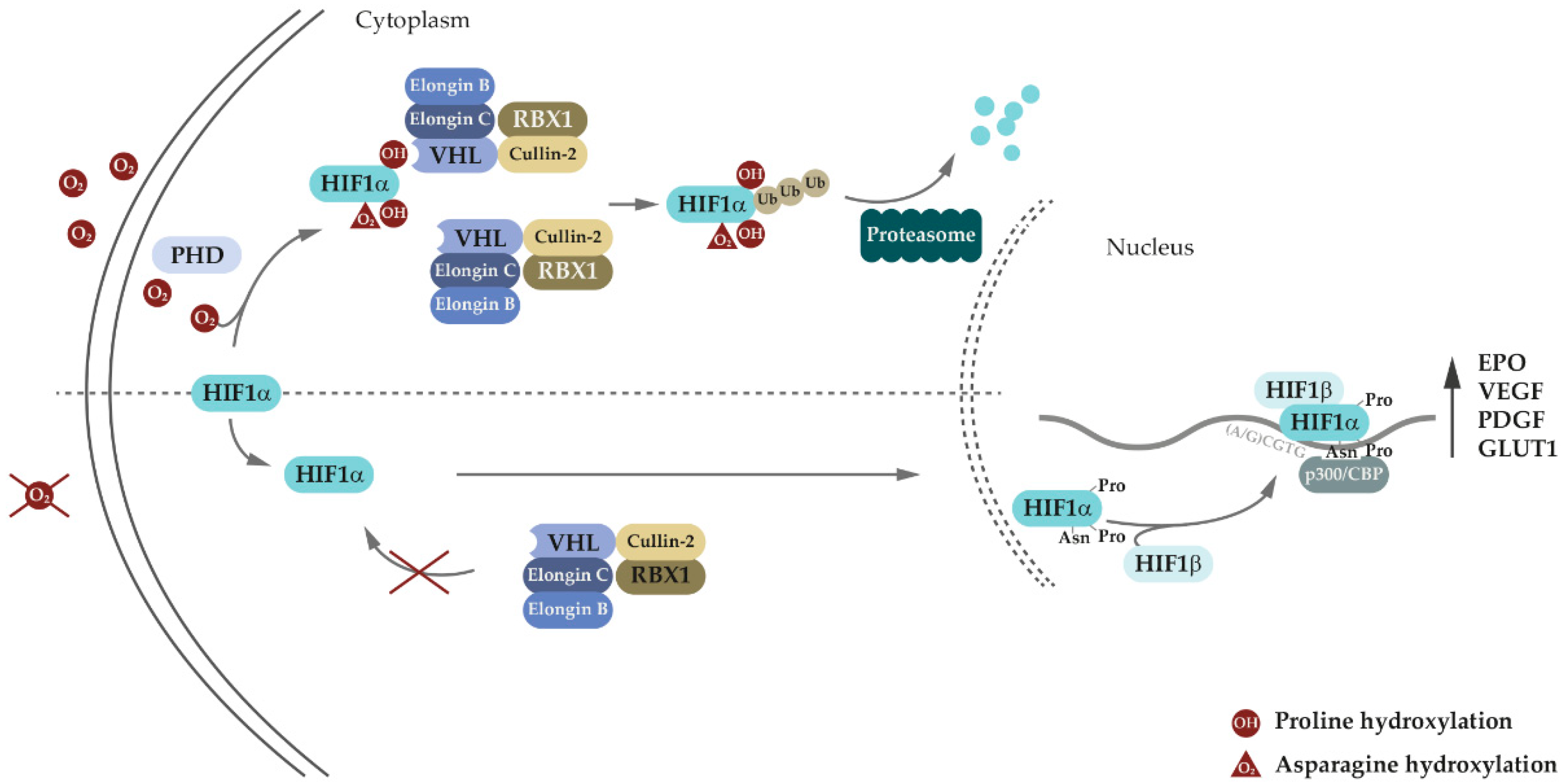

3. Genetic and Molecular Basis of VHL Disease
| Variant | Protein Change | Codon | VHL Type/Phenotype | Functional Consequence | Reference |
|---|---|---|---|---|---|
| c.191G>C | R64P | 64 | Type 2C | Increased aPKC JUNB levels; impaired binding to fibronectin. | [87][88] |
| c.194C>T | S65L | 56 | Type 2B | Impaired HIF1α binding; impaired HIF2α regulation. | [89][90][91] |
| c.208G>A | E70K | 70 | Type 1 | Impaired HIF1α binding. | [92][93] |
| c.233A>G | N78S | 78 | Type 1 | Impaired HIF1α regulation. | [94][95] |
| c.239G>A | S80N | 80 | Type 2C | No known consequence. | [94] |
| c.245G>C | R82P | 82 | Type 2B | Loss of function of VHL. | [96] |
| c.250G>C | V84L | 84 | Type 2C | No known consequence. | [94] |
| c.262T>A c.262T>C |
W88R | 88 | Hemangio- Blastoma 1 |
No known consequence. | [90] |
| c.269A>T | N90I | 90 | Type 2B | Impaired HIF1α regulation. | [90][94][97][98] |
| c.292T>C | Y98H | 98 | Type 2A | Impaired HIF1α regulation; defective microtubule stabilization. | [87][94] |
| c.292T>A | Y98N | 98 | Type 2B | Impaired HIF1α regulation; impaired GLUT1 suppression. | [97] |
| c.334T>A | Y112H | 112 | Type 2A | Impaired HIF1α regulation; decreased VHL stability. | [85][99] |
| c.334T>A | Y112N | 112 | Type 2B | Reduced stability of the Vhl-Elongin B/C complex; impaired HIF1α regulation; elevated HIF2α, GLUT1, and cyclin D1 expression in normoxic conditions. | [99][100][101] |
| c.334T>G | Y112D | 112 | Type 2C | No known consequence. | [94] |
| c.340G>C | G114R | 114 | Type 2B | Reduced stability of the Vhl-Elongin B/C complex. | [102] |
| c.349T>C c.349T>A |
W117R | 117 | Type 2B | Impaired HIF1α regulation; impaired binding to fibronectin; elevated HIF2α and GLUT1 expression in normoxic conditions. | [62][101][103] |
| c.355T>C c.357C>G c.357C>A |
F119L | 119 | Type 2B | Decreased VHL stability; impaired HIF1α regulation. | [104] |
| c.407T>C | F136S | 136 | Type 2B | No known consequence. | [94] |
| c.407T>A | F136Y | 136 | Type 2B | No known consequence. | [94] |
| c.408T>G | F136L | 136 | Type 2B | Decreased VHL stability; impaired HIF1α regulation. | [104][105] |
| c.482G>C | R161P | 161 | Type 2B | Reduced stability of the Vhl-Elongin B/C complex; defective microtubule stabilization. | [106][107] |
| c.482G>A | R161Q | 161 | Type 2A; Type 2B | Reduced VHL stability. | [108] |
| c.486C>G | C162W | 162 | Hemangio- Blastoma 1 |
Impaired HIF1α regulation. | [90][109] |
| c.499C>T | R167W | 167 | Type 2B | Decreased binding to Elongin B/C and Cullin-2; impaired ubiquitination and degradation of ESR1. | [62][110] |
| c.500G>A | R167Q | 167 | Hemangio- Blastoma 1 |
Decreased binding to Elongin C; impaired HIF2α regulation. | [90][111][112] |
| c.562C>G | L188V | Type 2C | Impaired binding to fibronectin; elevated RWWD3, aPKC, and JUNB levels. | [87][88][113][114] |
References
- Chou, A.; Toon, C.; Pickett, J.; Gill, A.J. von Hippel-Lindau syndrome. Front. Horm. Res. 2013, 41, 30–49.
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau disease. Handb. Clin. Neurol. 2015, 132, 139–156.
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067.
- Ding, X.; Zhang, C.; Frerich, J.M.; Germanwala, A.; Yang, C.; Lonser, R.R.; Mao, Y.; Zhuang, Z.; Zhang, M. De novo VHL germline mutation detected in a patient with mild clinical phenotype of von Hippel-Lindau disease. J. Neurosurg. 2014, 121, 384–386.
- Glasker, S.; Sohn, T.S.; Okamoto, H.; Li, J.; Lonser, R.R.; Oldfield, E.H.; Vortmeyer, A.O.; Zhuang, Z. Second hit deletion size in von Hippel-Lindau disease. Ann. Neurol. 2006, 59, 105–110.
- Murro, V.; Lippera, M.; Mucciolo, D.P.; Canu, L.; Ercolino, T.; De Filpo, G.; Giorgio, D.; Traficante, G.; Sodi, A.; Virgili, G.; et al. Outcome and genetic analysis of patients affected by retinal capillary hemangioblastoma in von Hippel Lindau syndrome. Mol. Vis. 2021, 27, 542–554.
- Richards, F.M.; Payne, S.J.; Zbar, B.; Affara, N.A.; Ferguson-Smith, M.A.; Maher, E.R. Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene. Hum. Mol. Genet. 1995, 4, 2139–2143.
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621.
- Ong, K.R.; Woodward, E.R.; Killick, P.; Lim, C.; Macdonald, F.; Maher, E.R. Genotype-phenotype correlations in von Hippel-Lindau disease. Hum. Mutat. 2007, 28, 143–149.
- Woodward, E.R.; Eng, C.; McMahon, R.; Voutilainen, R.; Affara, N.A.; Ponder, B.A.; Maher, E.R. Genetic predisposition to phaeochromocytoma: Analysis of candidate genes GDNF, RET and VHL. Hum. Mol. Genet. 1997, 6, 1051–1056.
- Bento, C.; Percy, M.J.; Gardie, B.; Maia, T.M.; van Wijk, R.; Perrotta, S.; Della Ragione, F.; Almeida, H.; Rossi, C.; Girodon, F.; et al. Genetic basis of congenital erythrocytosis: Mutation update and online databases. Hum. Mutat. 2014, 35, 15–26.
- Aronow, M.E.; Wiley, H.E.; Gaudric, A.; Krivosic, V.; Gorin, M.B.; Shields, C.L.; Shields, J.A.; Jonasch, E.W.; Singh, A.D.; Chew, E.Y. VON HIPPEL-LINDAU DISEASE: Update on Pathogenesis and Systemic Aspects. Retina 2019, 39, 2243–2253.
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320.
- Blankenship, C.; Naglich, J.G.; Whaley, J.M.; Seizinger, B.; Kley, N. Alternate choice of initiation codon produces a biologically active product of the von Hippel Lindau gene with tumor suppressor activity. Oncogene 1999, 18, 1529–1535.
- Lenglet, M.; Robriquet, F.; Schwarz, K.; Camps, C.; Couturier, A.; Hoogewijs, D.; Buffet, A.; Knight, S.J.L.; Gad, S.; Couve, S.; et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 2018, 132, 469–483.
- Iliopoulos, O.; Ohh, M.; Kaelin, W.G., Jr. pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc. Natl. Acad. Sci. USA 1998, 95, 11661–11666.
- Richards, F.M.; Schofield, P.N.; Fleming, S.; Maher, E.R. Expression of the von Hippel-Lindau disease tumour suppressor gene during human embryogenesis. Hum. Mol. Genet. 1996, 5, 639–644.
- Schoenfeld, A.; Davidowitz, E.J.; Burk, R.D. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc. Natl. Acad. Sci. USA 1998, 95, 8817–8822.
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745.
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489.
- Min, J.H.; Yang, H.; Ivan, M.; Gertler, F.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of an HIF-1alpha -pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889.
- Stebbins, C.E.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461.
- Leonardi, E.; Murgia, A.; Tosatto, S.C. Adding structural information to the von Hippel-Lindau (VHL) tumor suppressor interaction network. FEBS Lett. 2009, 583, 3704–3710.
- Okuda, H.; Hirai, S.; Takaki, Y.; Kamada, M.; Baba, M.; Sakai, N.; Kishida, T.; Kaneko, S.; Yao, M.; Ohno, S.; et al. Direct interaction of the β-domain of VHL tumor suppressor protein with the regulatory domain of atypical PKC isotypes. Biochem. Biophys. Res. Commun. 1999, 263, 491–497.
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30.
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406.
- Wang, D.; Eraslan, B.; Wieland, T.; Hallstrom, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503.
- Perl, K.; Ushakov, K.; Pozniak, Y.; Yizhar-Barnea, O.; Bhonker, Y.; Shivatzki, S.; Geiger, T.; Avraham, K.B.; Shamir, R. Reduced changes in protein compared to mRNA levels across non-proliferating tissues. BMC Genom. 2017, 18, 305.
- Hsu, T. Complex cellular functions of the von Hippel-Lindau tumor suppressor gene: Insights from model organisms. Oncogene 2012, 31, 2247–2257.
- Li, T.; Mao, C.; Wang, X.; Shi, Y.; Tao, Y. Epigenetic crosstalk between hypoxia and tumor driven by HIF regulation. J. Exp. Clin. Cancer Res. 2020, 39, 224.
- Liao, C.; Zhang, Q. Understanding the Oxygen-Sensing Pathway and Its Therapeutic Implications in Diseases. Am. J. Pathol. 2020, 190, 1584–1595.
- Tomc, J.; Debeljak, N. Molecular Insights into the Oxygen-Sensing Pathway and Erythropoietin Expression Regulation in Erythropoiesis. Int. J. Mol. Sci. 2021, 22, 7074.
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682.
- Iliopoulos, O.; Kibel, A.; Gray, S.; Kaelin, W.G., Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat. Med. 1995, 1, 822–826.
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 1995, 269, 1444–1446.
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237.
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G., Jr.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599.
- Lonergan, K.M.; Iliopoulos, O.; Ohh, M.; Kamura, T.; Conaway, R.C.; Conaway, J.W.; Kaelin, W.G., Jr. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell Biol. 1998, 18, 732–741.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275.
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468.
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472.
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15.
- Metzen, E.; Ratcliffe, P.J. HIF hydroxylation and cellular oxygen sensing. Biol. Chem. 2004, 385, 223–230.
- Hirsila, M.; Koivunen, P.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780.
- Chan, D.A.; Sutphin, P.D.; Yen, S.E.; Giaccia, A.J. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 α. Mol. Cell Biol. 2005, 25, 6415–6426.
- Tian, Y.M.; Yeoh, K.K.; Lee, M.K.; Eriksson, T.; Kessler, B.M.; Kramer, H.B.; Edelmann, M.J.; Willam, C.; Pugh, C.W.; Schofield, C.J.; et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J. Biol. Chem. 2011, 286, 13041–13051.
- He, W.; Batty-Stuart, S.; Lee, J.E.; Ohh, M. HIF-1alpha Hydroxyprolines Modulate Oxygen-Dependent Protein Stability Via Single VHL Interface With Comparable Effect on Ubiquitination Rate. J. Mol. Biol. 2021, 433, 167244.
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 2002, 277, 26351–26355.
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861.
- Zhou, M.I.; Wang, H.; Ross, J.J.; Kuzmin, I.; Xu, C.; Cohen, H.T. The von Hippel-Lindau tumor suppressor stabilizes novel plant homeodomain protein Jade-1. J. Biol. Chem. 2002, 277, 39887–39898.
- Zeng, L.; Bai, M.; Mittal, A.K.; El-Jouni, W.; Zhou, J.; Cohen, D.M.; Zhou, M.I.; Cohen, H.T. Candidate tumor suppressor and pVHL partner Jade-1 binds and inhibits AKT in renal cell carcinoma. Cancer Res. 2013, 73, 5371–5380.
- Schermer, B.; Ghenoiu, C.; Bartram, M.; Muller, R.U.; Kotsis, F.; Hohne, M.; Kuhn, W.; Rapka, M.; Nitschke, R.; Zentgraf, H.; et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J. Cell Biol. 2006, 175, 547–554.
- Frew, I.J.; Smole, Z.; Thoma, C.R.; Krek, W. Genetic deletion of the long isoform of the von Hippel-Lindau tumour suppressor gene product alters microtubule dynamics. Eur. J. Cancer 2013, 49, 2433–2440.
- Thoma, C.R.; Toso, A.; Gutbrodt, K.L.; Reggi, S.P.; Frew, I.J.; Schraml, P.; Hergovich, A.; Moch, H.; Meraldi, P.; Krek, W. VHL loss causes spindle misorientation and chromosome instability. Nat. Cell Biol. 2009, 11, 994–1001.
- Hsu, T.; Adereth, Y.; Kose, N.; Dammai, V. Endocytic function of von Hippel-Lindau tumor suppressor protein regulates surface localization of fibroblast growth factor receptor 1 and cell motility. J. Biol. Chem. 2006, 281, 12069–12080.
- Jinesh, G.G.; Kamat, A.M. RalBP1 and p19-VHL play an oncogenic role, and p30-VHL plays a tumor suppressor role during the blebbishield emergency program. Cell Death Discov. 2017, 3, 17023.
- Yang, H.; Minamishima, Y.A.; Yan, Q.; Schlisio, S.; Ebert, B.L.; Zhang, X.; Zhang, L.; Kim, W.Y.; Olumi, A.F.; Kaelin, W.G., Jr. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol. Cell. 2007, 28, 15–27.
- Kuznetsova, A.V.; Meller, J.; Schnell, P.O.; Nash, J.A.; Ignacak, M.L.; Sanchez, Y.; Conaway, J.W.; Conaway, R.C.; Czyzyk-Krzeska, M.F. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc. Natl. Acad. Sci. USA 2003, 100, 2706–2711.
- Mikhaylova, O.; Ignacak, M.L.; Barankiewicz, T.J.; Harbaugh, S.V.; Yi, Y.; Maxwell, P.H.; Schneider, M.; Van Geyte, K.; Carmeliet, P.; Revelo, M.P.; et al. The von Hippel-Lindau tumor suppressor protein and Egl-9-Type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol. Cell Biol. 2008, 28, 2701–2717.
- Na, X.; Duan, H.O.; Messing, E.M.; Schoen, S.R.; Ryan, C.K.; di Sant’Agnese, P.A.; Golemis, E.A.; Wu, G. Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel-Lindau protein. EMBO J. 2003, 22, 4249–4259.
- Calzada, M.J.; Esteban, M.A.; Feijoo-Cuaresma, M.; Castellanos, M.C.; Naranjo-Suarez, S.; Temes, E.; Mendez, F.; Yanez-Mo, M.; Ohh, M.; Landazuri, M.O. von Hippel-Lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res. 2006, 66, 1553–1560.
- Ohh, M.; Yauch, R.L.; Lonergan, K.M.; Whaley, J.M.; Stemmer-Rachamimov, A.O.; Louis, D.N.; Gavin, B.J.; Kley, N.; Kaelin, W.G., Jr.; Iliopoulos, O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol. Cell 1998, 1, 959–968.
- Sevilla-Montero, J.; Bienes-Martinez, R.; Labrousse-Arias, D.; Fuertes-Yebra, E.; Ordonez, A.; Calzada, M.J. pVHL-mediated regulation of the anti-angiogenic protein thrombospondin-1 decreases migration of Clear Cell Renal Carcinoma Cell Lines. Sci. Rep. 2020, 10, 1175.
- Tang, N.; Mack, F.; Haase, V.H.; Simon, M.C.; Johnson, R.S. pVHL function is essential for endothelial extracellular matrix deposition. Mol. Cell Biol. 2006, 26, 2519–2530.
- Danilin, S.; Sourbier, C.; Thomas, L.; Rothhut, S.; Lindner, V.; Helwig, J.J.; Jacqmin, D.; Lang, H.; Massfelder, T. von Hippel-Lindau tumor suppressor gene-dependent mRNA stabilization of the survival factor parathyroid hormone-related protein in human renal cell carcinoma by the RNA-binding protein HuR. Carcinogenesis 2009, 30, 387–396.
- Galban, S.; Martindale, J.L.; Mazan-Mamczarz, K.; Lopez de Silanes, I.; Fan, J.; Wang, W.; Decker, J.; Gorospe, M. Influence of the RNA-binding protein HuR in pVHL-regulated p53 expression in renal carcinoma cells. Mol. Cell Biol. 2003, 23, 7083–7095.
- Xin, H.; Brown, J.A.; Gong, C.; Fan, H.; Brewer, G.; Gnarra, J.R. Association of the von Hippel-Lindau protein with AUF1 and posttranscriptional regulation of VEGFA mRNA. Mol. Cancer Res. 2012, 10, 108–120.
- Kim, W.Y.; Sharpless, N.E. VHL inactivation: A new road to senescence. Cancer Cell 2008, 13, 295–297.
- Young, A.P.; Schlisio, S.; Minamishima, Y.A.; Zhang, Q.; Li, L.; Grisanzio, C.; Signoretti, S.; Kaelin, W.G., Jr. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol. 2008, 10, 361–369.
- Metcalf, J.L.; Bradshaw, P.S.; Komosa, M.; Greer, S.N.; Stephen Meyn, M.; Ohh, M. K63-ubiquitylation of VHL by SOCS1 mediates DNA double-strand break repair. Oncogene 2014, 33, 1055–1065.
- Russell, R.C.; Sufan, R.I.; Zhou, B.; Heir, P.; Bunda, S.; Sybingco, S.S.; Greer, S.N.; Roche, O.; Heathcote, S.A.; Chow, V.W.; et al. Loss of JAK2 regulation via a heterodimeric VHL-SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat. Med. 2011, 17, 845–853.
- Lai, Y.; Song, M.; Hakala, K.; Weintraub, S.T.; Shiio, Y. Proteomic dissection of the von Hippel-Lindau (VHL) interactome. J. Proteome Res. 2011, 10, 5175–5182.
- Minervini, G.; Mazzotta, G.M.; Masiero, A.; Sartori, E.; Corra, S.; Potenza, E.; Costa, R.; Tosatto, S.C. Isoform-specific interactions of the von Hippel-Lindau tumor suppressor protein. Sci. Rep. 2015, 5, 12605.
- Jansson, M.; Durant, S.T.; Cho, E.C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439.
- Roe, J.S.; Kim, H.; Lee, S.M.; Kim, S.T.; Cho, E.J.; Youn, H.D. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 2006, 22, 395–405.
- Kong, L.; Yin, H.; Yuan, L. Centrosomal MCM7 strengthens the Cep68-VHL interaction and excessive MCM7 leads to centrosome splitting resulting from increase in Cep68 ubiquitination and proteasomal degradation. Biochem. Biophys. Res. Commun. 2017, 489, 497–502.
- Hasanov, E.; Chen, G.; Chowdhury, P.; Weldon, J.; Ding, Z.; Jonasch, E.; Sen, S.; Walker, C.L.; Dere, R. Ubiquitination and regulation of AURKA identifies a hypoxia-independent E3 ligase activity of VHL. Oncogene 2017, 36, 3450–3463.
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613.
- Choi, H.; Kim, K.J.; Hong, N.; Shin, S.; Choi, J.R.; Kang, S.W.; Lee, S.T.; Rhee, Y. Genetic Analysis and Clinical Characteristics of Hereditary Pheochromocytoma and Paraganglioma Syndrome in Korean Population. Endocrinol. Metab. 2020, 35, 858–872.
- Tabaro, F.; Minervini, G.; Sundus, F.; Quaglia, F.; Leonardi, E.; Piovesan, D.; Tosatto, S.C. VHLdb: A database of von Hippel-Lindau protein interactors and mutations. Sci. Rep. 2016, 6, 31128.
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404.
- Maher, E.R.; Sandford, R.N. von Hippel-Lindau Disease: An Update. Curr. Genet. Med. Rep. 2019, 7, 227–235.
- Crespigio, J.; Berbel, L.C.L.; Dias, M.A.; Berbel, R.F.; Pereira, S.S.; Pignatelli, D.; Mazzuco, T.L. Von Hippel-Lindau disease: A single gene, several hereditary tumors. J. Endocrinol. Investig. 2018, 41, 21–31.
- Hacker, K.E.; Lee, C.M.; Rathmell, W.K. VHL type 2B mutations retain VBC complex form and function. PLoS ONE 2008, 3, e3801.
- Buart, S.; Terry, S.; Diop, M.K.; Dessen, P.; Couve, S.; Abdou, A.; Adam, J.; Thiery, J.; Savagner, P.; Chouaib, S. The Most Common VHL Point Mutation R167Q in Hereditary VHL Disease Interferes with Cell Plasticity Regulation. Cancers 2021, 13, 3897.
- Hoffman, M.A.; Ohh, M.; Yang, H.; Klco, J.M.; Ivan, M.; Kaelin, W.G., Jr. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum. Mol. Genet. 2001, 10, 1019–1027.
- Lee, S.; Nakamura, E.; Yang, H.; Wei, W.; Linggi, M.S.; Sajan, M.P.; Farese, R.V.; Freeman, R.S.; Carter, B.D.; Kaelin, W.G., Jr.; et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell 2005, 8, 155–167.
- Guo, K.; Wei, Y.; Wang, Z.; Zhang, X.; Zhang, X.; Liu, X.; Wu, W.; Wu, Z.; Zhang, L.; Cui, C.P. Deubiquitylase OTUD6B stabilizes the mutated pVHL and suppresses cell migration in clear cell renal cell carcinoma. Cell Death Dis. 2022, 13, 97.
- Hong, B.; Ma, K.; Zhou, J.; Zhang, J.; Wang, J.; Liu, S.; Zhang, Z.; Cai, L.; Zhang, N.; Gong, K. Frequent Mutations of VHL Gene and the Clinical Phenotypes in the Largest Chinese Cohort With Von Hippel-Lindau Disease. Front. Genet. 2019, 10, 867.
- Miller, F.; Kentsis, A.; Osman, R.; Pan, Z.Q. Inactivation of VHL by tumorigenic mutations that disrupt dynamic coupling of the pVHL.hypoxia-inducible transcription factor-1alpha complex. J. Biol. Chem. 2005, 280, 7985–7996.
- Forman, J.R.; Worth, C.L.; Bickerton, G.R.; Eisen, T.G.; Blundell, T.L. Structural bioinformatics mutation analysis reveals genotype-phenotype correlations in von Hippel-Lindau disease and suggests molecular mechanisms of tumorigenesis. Proteins 2009, 77, 84–96.
- Hwang, S.; Ku, C.R.; Lee, J.I.; Hur, K.Y.; Lee, M.S.; Lee, C.H.; Koo, K.Y.; Lee, J.S.; Rhee, Y. Germline mutation of Glu70Lys is highly frequent in Korean patients with von Hippel-Lindau (VHL) disease. J. Hum. Genet. 2014, 59, 488–493.
- Hergovich, A.; Lisztwan, J.; Barry, R.; Ballschmieter, P.; Krek, W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat. Cell Biol. 2003, 5, 64–70.
- Lin, G.; Zhao, Y.; Zhang, Z.; Zhang, H. Clinical diagnosis, treatment and screening of the VHL gene in three von Hippel-Lindau disease pedigrees. Exp. Ther. Med. 2020, 20, 1237–1244.
- Li, Z.; Na, X.; Wang, D.; Schoen, S.R.; Messing, E.M.; Wu, G. Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2002, 277, 4656–4662.
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741.
- Vaux, E.C.; Wood, S.M.; Cockman, M.E.; Nicholls, L.G.; Yeates, K.M.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J. Selection of mutant CHO cells with constitutive activation of the HIF system and inactivation of the von Hippel-Lindau tumor suppressor. J. Biol. Chem. 2001, 276, 44323–44330.
- Knauth, K.; Bex, C.; Jemth, P.; Buchberger, A. Renal cell carcinoma risk in type 2 von Hippel-Lindau disease correlates with defects in pVHL stability and HIF-1alpha interactions. Oncogene 2006, 25, 370–377.
- Chung, J.; Roberts, A.M.; Chow, J.; Coady-Osberg, N.; Ohh, M. Homotypic association between tumour-associated VHL proteins leads to the restoration of HIF pathway. Oncogene 2006, 25, 3079–3083.
- Li, L.; Zhang, L.; Zhang, X.; Yan, Q.; Minamishima, Y.A.; Olumi, A.F.; Mao, M.; Bartz, S.; Kaelin, W.G., Jr. Hypoxia-inducible factor linked to differential kidney cancer risk seen with type 2A and type 2B VHL mutations. Mol. Cell Biol. 2007, 27, 5381–5392.
- Feldman, D.E.; Spiess, C.; Howard, D.E.; Frydman, J. Tumorigenic mutations in VHL disrupt folding in vivo by interfering with chaperonin binding. Mol. Cell 2003, 12, 1213–1224.
- Hansen, W.J.; Ohh, M.; Moslehi, J.; Kondo, K.; Kaelin, W.G.; Welch, W.J. Diverse effects of mutations in exon II of the von Hippel-Lindau (VHL) tumor suppressor gene on the interaction of pVHL with the cytosolic chaperonin and pVHL-dependent ubiquitin ligase activity. Mol. Cell Biol. 2002, 22, 1947–1960.
- Shmueli, M.D.; Schnaider, L.; Rosenblum, D.; Herzog, G.; Gazit, E.; Segal, D. Structural insights into the folding defects of oncogenic pVHL lead to correction of its function in vitro. PLoS ONE 2013, 8, e66333.
- Shmueli, M.D.; Levy-Kanfo, L.; Haj, E.; Schoenfeld, A.R.; Gazit, E.; Segal, D. Arginine refolds, stabilizes, and restores function of mutant pVHL proteins in animal model of the VHL cancer syndrome. Oncogene 2019, 38, 1038–1049.
- Ohh, M.; Takagi, Y.; Aso, T.; Stebbins, C.E.; Pavletich, N.P.; Zbar, B.; Conaway, R.C.; Conaway, J.W.; Kaelin, W.G., Jr. Synthetic peptides define critical contacts between elongin C, elongin B, and the von Hippel-Lindau protein. J. Clin. Investig. 1999, 104, 1583–1591.
- Thoma, C.R.; Matov, A.; Gutbrodt, K.L.; Hoerner, C.R.; Smole, Z.; Krek, W.; Danuser, G. Quantitative image analysis identifies pVHL as a key regulator of microtubule dynamic instability. J. Cell Biol. 2010, 190, 991–1003.
- Couve, S.; Ladroue, C.; Laine, E.; Mahtouk, K.; Guegan, J.; Gad, S.; Le Jeune, H.; Le Gentil, M.; Nuel, G.; Kim, W.Y.; et al. Genetic evidence of a precisely tuned dysregulation in the hypoxia signaling pathway during oncogenesis. Cancer Res. 2014, 74, 6554–6564.
- Iturrioz, X.; Parker, P.J. PKCzetaII is a target for degradation through the tumour suppressor protein pVHL. FEBS Lett. 2007, 581, 1397–1402.
- Jung, Y.S.; Lee, S.J.; Yoon, M.H.; Ha, N.C.; Park, B.J. Estrogen receptor α is a novel target of the Von Hippel-Lindau protein and is responsible for the proliferation of VHL-deficient cells under hypoxic conditions. Cell Cycle 2012, 11, 4462–4473.
- Arreola, A.; Payne, L.B.; Julian, M.H.; de Cubas, A.A.; Daniels, A.B.; Taylor, S.; Zhao, H.; Darden, J.; Bautch, V.L.; Rathmell, W.K.; et al. Von Hippel-Lindau mutations disrupt vascular patterning and maturation via Notch. JCI Insight 2018, 3, e92193.
- Rathmell, W.K.; Hickey, M.M.; Bezman, N.A.; Chmielecki, C.A.; Carraway, N.C.; Simon, M.C. In vitro and in vivo models analyzing von Hippel-Lindau disease-specific mutations. Cancer Res. 2004, 64, 8595–8603.
- Kurban, G.; Hudon, V.; Duplan, E.; Ohh, M.; Pause, A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 2006, 66, 1313–1319.
- Tedesco, L.; Elguero, B.; Pacin, D.G.; Senin, S.; Pollak, C.; Garcia Marchinena, P.A.; Jurado, A.M.; Isola, M.; Labanca, M.J.; Palazzo, M.; et al. von Hippel-Lindau mutants in renal cell carcinoma are regulated by increased expression of RSUME. Cell Death Dis. 2019, 10, 266.
- de Rojas, P.I.; Albinana, V.; Taranets, L.; Recio-Poveda, L.; Cuesta, A.M.; Popov, N.; Kronenberger, T.; Botella, L.M. The Endothelial Landscape and Its Role in Von Hippel-Lindau Disease. Cells 2021, 10, 2313.
- Glasker, S.; Vergauwen, E.; Koch, C.A.; Kutikov, A.; Vortmeyer, A.O. Von Hippel-Lindau Disease: Current Challenges and Future Prospects. Onco Targets Ther. 2020, 13, 5669–5690.
- Creighton, C.J.; Morgan, M.; Gunaratne, P.H.; Wheeler, D.A.; Gibbs, R.A.; Gordon Robertson, A.; Chu, A.; Beroukhim, R.; Cibulskis, K.; Signoretti, S.; et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49.
- Krieg, M.; Marti, H.H.; Plate, K.H. Coexpression of erythropoietin and vascular endothelial growth factor in nervous system tumors associated with von Hippel-Lindau tumor suppressor gene loss of function. Blood 1998, 92, 3388–3393.
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6.
- Gordeuk, V.R.; Prchal, J.T. Vascular complications in Chuvash polycythemia. Semin. Thromb. Hemost. 2006, 32, 289–294.
- Kaelin, W.G., Jr. The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem. Biophys. Res. Commun. 2005, 338, 627–638.
- Berger, A.H.; Knudson, A.G.; Pandolfi, P.P. A continuum model for tumour suppression. Nature 2011, 476, 163–169.
- Ang, S.O.; Chen, H.; Gordeuk, V.R.; Sergueeva, A.I.; Polyakova, L.A.; Miasnikova, G.Y.; Kralovics, R.; Stockton, D.W.; Prchal, J.T. Endemic polycythemia in Russia: Mutation in the VHL gene. Blood Cells Mol. Dis. 2002, 28, 57–62.
- Polyakova, L.A. Familial erythrocytosis among inhabitants of the Chuvash ASSR. Probl. Gematol. Pereliv. Krovi 1974, 10, 30–36.
- Sergeyeva, A.; Gordeuk, V.R.; Tokarev, Y.N.; Sokol, L.; Prchal, J.F.; Prchal, J.T. Congenital polycythemia in Chuvashia. Blood 1997, 89, 2148–2154.