Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Andreea-Adriana Neamțu and Version 3 by Lindsay Dong.
Quercetin is a polyphenolic flavonoid plant secondary metabolite with a well-characterized antioxidant activity. It has been extensively reported as an anti-carcinogenic agent, and the modulated targets of quercetin have been also characterized in the context of colorectal cancer (CRC).
- quercetin
- flavonol
- polyphenol
- colorectal cancer
1. Quercetin
Quercetin is a plant pigment and secondary metabolite, a polyphenolic flavonoid phytochemical with a well-characterized antioxidant activity [1][2]. Chemically, it is a pentahydroxyflavone, using as a backbone the flavone structure C6(A-ring)-C3(C-ring)-C6(B-ring) (
Quercetin is a plant pigment and secondary metabolite, a polyphenolic flavonoid phytochemical with a well-characterized antioxidant activity [21,22]. Chemically, it is a pentahydroxyflavone, using as a backbone the flavone structure C6(A-ring)-C3(C-ring)-C6(B-ring) (
Figure 1) [2]. The official IUPAC name of the compound is 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one and the chemical structure C
) [22]. The official IUPAC name of the compound is 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one and the chemical structure C
15
H
10
O
7 [3][4]. The molecular weight of quercetin is 302,2 g/mol, and in its purified form it is a yellow-colored crystalline solid at room temperature, with poor water solubility, but increased solubility in alkaline aqueous solutions and alcohols, having a low acute toxicity level through oral exposure at LD
[23,24]. The molecular weight of quercetin is 302,2 g/mol, and in its purified form it is a yellow-colored crystalline solid at room temperature, with poor water solubility, but increased solubility in alkaline aqueous solutions and alcohols, having a low acute toxicity level through oral exposure at LD
50 161 mg/kg [2][5]. The average daily intake is approximately 25 mg according to the US Department of Health and Human Services and studies carried out in Japan, France, and Finland [6][7][8][9][10], due to consumption of major food sources such as onions, asparagus, and berries, while reduced quantities are acquired from various other plants (
161 mg/kg [22,25]. The average daily intake is approximately 25 mg according to the US Department of Health and Human Services and studies carried out in Japan, France, and Finland [26,27,28,29,30], due to consumption of major food sources such as onions, asparagus, and berries, while reduced quantities are acquired from various other plants (
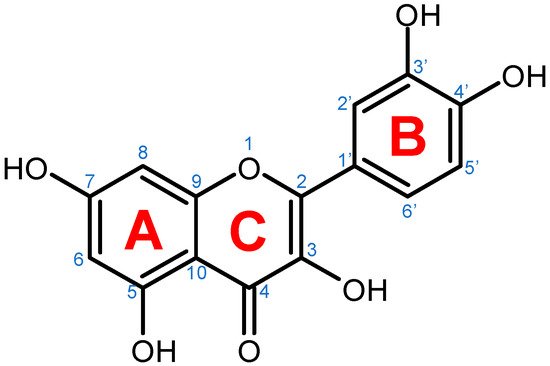
Figure 1.
Chemical structure of quercetin with numbered carbon atoms (blue) and marked rings (red) on the general flavonoid backbone structure.
Table 1.
Dietary sources with quercetin concentrations higher than 10 mg/100 g fresh weight according to the United States Department of Agriculture Database for the Flavonoid Content of Selected Foods.
| Plant | Quercetin Concentration |
|---|---|
| (mg/100 g Fresh Weight) |
| Dill |
| 79.0 |
| Fennel leaves | 46.8 |
| Onion | 45.0 |
| Oregano | 42.0 |
| Chili pepper | 32.6 |
| Spinach | 27.2 |
| Cranberry | 25.0 |
| Kale | 22.6 |
| Cherry | 17.4 |
| Lettuce | 14.7 |
| Blueberry | 14.6 |
| Asparagus | 14.0 |
| Broccoli | 13.7 |
| Chives | 10.4 |
Source: USDA (United States Department of Agriculture) Database for the Flavonoid Content of Selected Foods [32].
2. Quercetin and Its Derivatives—Mechanisms of Action in CRC
Quercetin and its derivatives act on a multitude of targets involved in the initiation and promotion/progression phases of colorectal cancer (CRC) carcinogenesis, as described in established cell lines and other animal models of the disease. Among the anti-carcinogenic activities of quercetin, the most notable described in CRC are inhibition of cellular proliferation and growth, cell cycle arrest, induction of apoptosis, reduction in tumor size, decrease in number of tumor nodule, suppression of metastasis, decrease in inflammation, decrease in ROS (i.e., antioxidant activity), and reduction in multidrug resistance. Well documented in cell culture and rodent studies, the mechanisms of action and targets of quercetin mainly involve members of the pathways Wnt/β-catenin, PI3K/AKT/mTOR, MAPK/Erk, MAPK/JNK, MAPK/p38, p-53, and NF-κB (Quercetin and its derivatives act on a multitude of targets involved in the initiation and promotion/progression phases of CRC carcinogenesis, as described in established cell lines and other animal models of the disease. Among the anti-carcinogenic activities of quercetin, the most notable described in CRC are inhibition of cellular proliferation and growth, cell cycle arrest, induction of apoptosis, reduction in tumor size, decrease in number of tumor nodule, suppression of metastasis, decrease in inflammation, decrease in ROS (i.e., antioxidant activity), and reduction in multidrug resistance. Well documented in cell culture and rodent studies, the mechanisms of action and targets of quercetin mainly involve members of the pathways Wnt/β-catenin, PI3K/AKT/mTOR, MAPK/Erk, MAPK/JNK, MAPK/p38, p-53, and NF-κB (
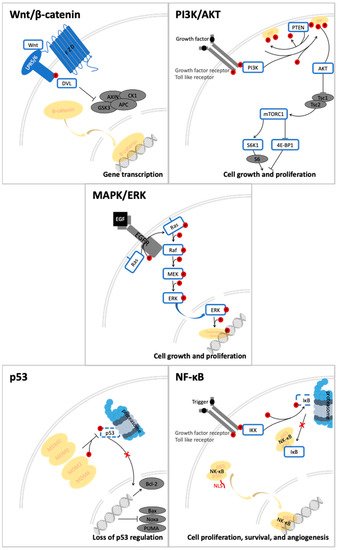
Figure 24.
Signal transduction pathways in CRC that are modulated by quercetin: Wnt/β-catenin, PI3K/AKT, MAPK (using MAPK/ERK as an example for the phosphorylation cascade), p53, and NF-κB.
3. Crucial Signal Transduction Pathways in CRC
3.1. Wnt/β-Catenin Signaling in CRC
The critical role of the Wnt/β-catenin pathway (3.1. Wnt/β-Catenin Signaling in CRC
The critical role of the Wnt/β-catenin pathway (
Figure 2) in the etiology of CRC has been thoroughly studied; thus, light has been shed on the molecular mechanisms of interaction and the signal transduction regulation. Genetic alterations in the members of Wnt/β-catenin pathway lead to the intrinsic aberrant canonical Wnt/β-catenin activation, mainly stemming from mutations in the APC, AXIN1, and AXIN2 genes [17].
Physiologically, β-catenin levels are maintained at sub-critical levels through the dynamic activity of the degradosome complex, consisting of the glycogen synthase kinase 3 (GSK3), axis inhibition protein 1 (AXIN1), adenomatous polyposis coli (APC), E3-ubiquitin ligase β-TrCP, protein phosphatase 2A (PP2A), and casein kinase 1α (CK1α) [18]. While the scaffold proteins in the complex are APC and AXIN1, the CK1α and GSK3 are responsible for β-catenin phosphorylation as serine/threonine kinases [19]. Once phosphorylated, E3-ubiquitin ligase β-TrCP mediates its ubiquitination and targets it for degradation by the proteasome machinery [17][20].
Pathologically, the Wnt ligands bind the 7-transmembrane receptor frizzled (FZD) family and its co-receptors low-density lipoprotein receptors 5 and 6 (LRP5/6) [21][22]. The ligand-receptor complex Wnt-FZD-LRP5/6 assembly with recruitment of the Dishevelled (DVL) adaptor by FZD facilitates the phosphorylation of LRP6 [23]. This leads to a cascade of molecular interactions including its association with AXIN1, their translocation to the plasma membrane, the dissociation of GSK3 from AXIN1 and APC, and concomitant stabilization of β-catenin by dephosphorylation [23][24]. Thereafter, the signalosome is assembled, a multiprotein complex that transduces Wnt signals, and the degradosome is disassembled leading to β-catenin accumulation in the cytosol and its subsequent nuclear translocation [23]. Nuclear β-catenin acts as a transcriptional activator inducing the transcription of target genes, among which are c-MYK [25] and AXIN2 [26], hereafter activating the oncogenic mechanisms [17].
3.2. PI3K/AKT-mTOR Signaling in CRC
Another relevant signal transduction pathway in CRC development and progression is the PI3K/AKT/mTOR cascade (4) in the etiology of CRC has been thoroughly studied; thus, light has been shed on the molecular mechanisms of interaction and the signal transduction regulation. Genetic alterations in the members of Wnt/β-catenin pathway lead to the intrinsic aberrant canonical Wnt/β-catenin activation, mainly stemming from mutations in the APC, AXIN1, and AXIN2 genes [78].
Physiologically, β-catenin levels are maintained at sub-critical levels through the dynamic activity of the degradosome complex, consisting of the glycogen synthase kinase 3 (GSK3), axis inhibition protein 1 (AXIN1), adenomatous polyposis coli (APC), E3-ubiquitin ligase β-TrCP, protein phosphatase 2A (PP2A), and casein kinase 1α (CK1α) [79]. While the scaffold proteins in the complex are APC and AXIN1, the CK1α and GSK3 are responsible for β-catenin phosphorylation as serine/threonine kinases [80]. Once phosphorylated, E3-ubiquitin ligase β-TrCP mediates its ubiquitination and targets it for degradation by the proteasome machinery [78,81].
Pathologically, the Wnt ligands bind the 7-transmembrane receptor frizzled (FZD) family and its co-receptors low-density lipoprotein receptors 5 and 6 (LRP5/6) [82,83]. The ligand-receptor complex Wnt-FZD-LRP5/6 assembly with recruitment of the Dishevelled (DVL) adaptor by FZD facilitates the phosphorylation of LRP6 [84]. This leads to a cascade of molecular interactions including its association with AXIN1, their translocation to the plasma membrane, the dissociation of GSK3 from AXIN1 and APC, and concomitant stabilization of β-catenin by dephosphorylation [84,85]. Thereafter, the signalosome is assembled, a multiprotein complex that transduces Wnt signals, and the degradosome is disassembled leading to β-catenin accumulation in the cytosol and its subsequent nuclear translocation [84]. Nuclear β-catenin acts as a transcriptional activator inducing the transcription of target genes, among which are c-MYK [86] and AXIN2 [87], hereafter activating the oncogenic mechanisms [78].
2.2. PI3K/AKT-mTOR Signaling in CRC
Another relevant signal transduction pathway in CRC development and progression is the PI3K/AKT/mTOR cascade (
Figure 2). It tightly interacts with the previously mentioned Wnt/β-catenin pathway, as blockage of, more accurately, PI3K/AKT/mTORC1 leads to hyperactivation of the Wnt/β-catenin as compensatory mechanism (
4). It tightly interacts with the previously mentioned Wnt/β-catenin pathway, as blockage of, more accurately, PI3K/AKT/mTORC1 leads to hyperactivation of the Wnt/β-catenin as compensatory mechanism (
) [78].
The serine/threonine protein kinase mTOR consists of two multiprotein complexes: mTORC1 and mTORC2 [88], with the regulatory-associated protein of mTOR and the proline-rich AKT substrate of 40 KDa (PRAS40) being distinctive for the mTORC1 complex [89,90] and the rapamycin-insensitive companion of mTOR, the protein observed with RICTOR 1/2, and the mammalian stress-activated protein kinase-interacting protein 1 being distinctive for the mTORC2 complex [91,92,93]. While the molecular functions of mTORC2 were not fully elucidated, mTORC1 has been attributed several functions, some of which are vital pivots in the development of CRC. The most relevant upstream regulators of mTORC1 suffering genetic alterations in the context of CRC are: PIK3CA gene-gain-of-function mutations [94,95], PTEN gene-inactivating mutations [95], or the STK11/LKB1 gene [96], which encodes for an mTORC1 repressor. CRC reported mutations in the mTOR genes themselves are not as ubiquitous, while in the same pathway, the most seldom are the AKT gene mutations [97].
The mitogenic stimuli PI3K and AKT are the main activators of mTORC1 in the pathological context of CRC [98]. PI3K enzyme catalyzes the conversion of phosphatidylinositol (3,4)-bisphosphate into phosphatidylinositol (3,4,5)-trisphosphate, therefore triggering the phosphorylation of AKT [99]. Then, the AKT-mediated phosphorylation of mTORC1 takes place. This entails the phosphorylation of two main downstream targets: the eukaryotic translation initiation factor 4E (eIF4E) binding protein 1 (4E-BP1) and the S6 kinase 1 [100,101]. It is followed by 4E-BP1 dissociation from eIF4E, leading to mRNA translation activation, whilst S6K1 activation facilitates the phosphorylation of the S6 ribosomal protein leading to initiation and elongation of translation [102].
3.3. MAPK Cascades in CRC
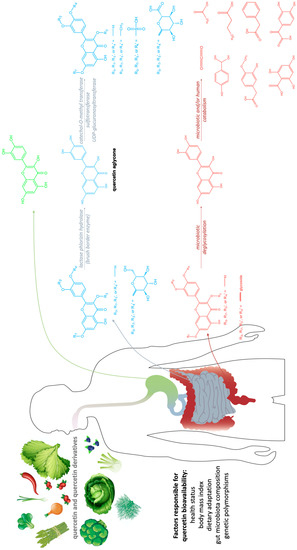
The mitogen-activated protein kinase (MAPK) cascades (Figure 3. Quercetin metabolism in the gastro-intestinal tract. Green—stomach absorption; blue—small intestine absorption; red—large bowel absorption.
The serine/threonine protein kinase mTOR consists of two multiprotein complexes: mTORC1 and mTORC2 [27], with the regulatory-associated protein of mTOR and the proline-rich AKT substrate of 40 KDa (PRAS40) being distinctive for the mTORC1 complex [28][29] and the rapamycin-insensitive companion of mTOR, the protein observed with RICTOR 1/2, and the mammalian stress-activated protein kinase-interacting protein 1 being distinctive for the mTORC2 complex [30][31][32]. While the molecular functions of mTORC2 were not fully elucidated, mTORC1 has been attributed several functions, some of which are vital pivots in the development of CRC. The most relevant upstream regulators of mTORC1 suffering genetic alterations in the context of CRC are: PIK3CA gene-gain-of-function mutations [33][34], PTEN gene-inactivating mutations [34], or the STK11/LKB1 gene [35], which encodes for an mTORC1 repressor. CRC reported mutations in the mTOR genes themselves are not as ubiquitous, while in the same pathway, the most seldom are the AKT gene mutations [36].
The mitogenic stimuli PI3K and AKT are the main activators of mTORC1 in the pathological context of CRC [37]. PI3K enzyme catalyzes the conversion of phosphatidylinositol (3,4)-bisphosphate into phosphatidylinositol (3,4,5)-trisphosphate, therefore triggering the phosphorylation of AKT [38]. Then, the AKT-mediated phosphorylation of mTORC1 takes place. This entails the phosphorylation of two main downstream targets: the eukaryotic translation initiation factor 4E (eIF4E) binding protein 1 (4E-BP1) and the S6 kinase 1 [39][40]. It is followed by 4E-BP1 dissociation from eIF4E, leading to mRNA translation activation, whilst S6K1 activation facilitates the phosphorylation of the S6 ribosomal protein leading to initiation and elongation of translation [41].
3.3. MAPK Cascades in CRC
The mitogen-activated protein kinase (MAPK) cascades (4) represent a set of membrane-to-nucleus signaling pathways that result in phosphorylation and activation of transcription factors [103]. With MAPK being members of the Ser/Thr kinases family, multiple rounds of subsequent phosphorylation-activating kinases are triggered [104].
There are three distinct MAPK cascades: MAPK/Erk (extracellular-signal-regulated kinases), MAPK/JNK (c-Jun N-terminal or stress-activated protein kinases), and MAPK/p38 [104]. As all pathways can be targeted (up- and down-regulated) by quercetin in the context of CRC, they will be the further discussed in the current publication.
Several proto-oncogenes are responsible for the involvement of the MAPK cascades in the development of CRC. The aberrations include the gain-of-function KRAS and BRAF gene mutations and upregulation of JUN gene [105]. Moreover, the role of EGFR is noteworthy in the MAPK activation and upregulation relevant in CRC [105].
3.3.1. MAPK/ERK Signaling in CRC
EGFR, a transmembrane protein, member of the ErbB family of receptors, functions as a receptor tyrosine kinase located upstream of MAPK pathways [104]. The three Ras small GTPases are H-Ras, N-Ras, and K-Ras [106], while the most relevant Raf kinases are A-Raf, B-Raf and C-Raf (Raf1) [107]. Through the phosphorylation of the inactive form of Ras-family GTPases bound to GDP to the active form bound to GTP, external signals are transmitted from receptors on the cytoplasmic membrane to the interior of the cell [108]. The adaptor complex then activates Ras-GTP. Post RAS activation, there is a phosphorylation-dependent cascade, activating RAF, MEK, and, finally, ERK. The activation of the ERK/MAPK pathway is reported to also induce the synthesis of cyclin D1, relevant in the progression of cell cycle [109]. Moreover, Raf1 creates a link between the MAPK/ERK pathway and PI3K/AKT, allowing the possibility of correlated feedback between the two distinct metabolic paths, both upregulated in the CRC pathology [105].
5.3.2. MAPK/JNK Signaling in CRC
The JNK cascade is used by the transforming growth factor-β (TGFβ) in order to autoregulate its concentration; however, it does not affect the JNK protein expression [110]. The pathway activation induced by TGFβ can act in conjunction with SMADs or in SMAD-independent manner [103,105]. The latter is acting on the MKK4–TGFβ-activated kinase 1 (TAK1) axis [110,111], where TAK1 is activated by the tumor necrosis factor-receptor-associated factor 6 (TRAF6), TGFβR2, and TGFβR1 protein complex [110]. Thereafter, it activates the JNK/p38 pathways [110], culminating in the formation of the ICD, domain of TGFβR1, consequent to ubiquitination and TNF-alpha converting enzyme cleavage [104]. ICD is able to translocate into the nucleus, where it induces the overexpression of Snail, MMP2, and p300 genes [112].
5.3.3. MAPK/ p38 Signaling in CRC
The MAPK/p38 pathway becomes activated in response to stressors such as hypoxia, heat shock, and osmotic shock [104]. p38 signaling is required for cell migration and metastasis in both CRC and breast cancer [113,114]. Similar to JNKs, p38 MAPKs are activated through autophosphorylation by MKKs [110,111]. TAK1 and TRAF6 are responsible for the SMAD-independent activation of p38. At this level, there is crosstalk in between TAK1 and the NF-κB-MMP9 pathway, another relevant carcinogenesis pathway reported in CRC. The blockade of p38 MAPK activity leads to the recovery of cell cycle and induction cell death mainly through autophagy [114,115].
5.4. p53 Signaling in CRC
TP53, a tumor suppressor gene, is among the most commonly mutated genes in CRC and various other types of cancer, with mutations mainly in the exons 5 to 8 (DNA binding domain) [116,117,118]. p53 is physiologically expressed at low levels, partly due to the negative feedback loops that involve MDM2. MDM2 is a transcriptional target of p53 that mediates the degradation of p53 through negative feedback and by functioning as an E3 ubiquitin-ligase that regulates the ubiquitination of p53 [119,120]. Low levels of p53 expression maintain homoeostasis of the cell cycle and cell death. A homolog of MDM2, namely, MDM4, not regulated by p53, forms heterodimers with MDM2 and can enhance MDM2 induced p53 degradation [119]. As a response to stress factors, such as oncogenes, DNA damage, UV irradiation, free radicals, hypoxia, or deficiencies in nutrients and growth factors, p53 is also activated (
Figure 2) represent a set of membrane-to-nucleus signaling pathways that result in phosphorylation and activation of transcription factors [42]. With MAPK being members of the Ser/Thr kinases family, multiple rounds of subsequent phosphorylation-activating kinases are triggered [43].
There are three distinct MAPK cascades: MAPK/Erk (extracellular-signal-regulated kinases), MAPK/JNK (c-Jun N-terminal or stress-activated protein kinases), and MAPK/p38 [43]. As all pathways can be targeted (up- and down-regulated) by quercetin in the context of CRC, they will be the further discussed in the current publication.
Several proto-oncogenes are responsible for the involvement of the MAPK cascades in the development of CRC. The aberrations include the gain-of-function KRAS and BRAF gene mutations and upregulation of JUN gene [44]. Moreover, the role of EGFR is noteworthy in the MAPK activation and upregulation relevant in CRC [44].
3.3.1. MAPK/ERK Signaling in CRC
EGFR, a transmembrane protein, member of the ErbB family of receptors, functions as a receptor tyrosine kinase located upstream of MAPK pathways [43]. The three Ras small GTPases are H-Ras, N-Ras, and K-Ras [45], while the most relevant Raf kinases are A-Raf, B-Raf and C-Raf (Raf1) [46]. Through the phosphorylation of the inactive form of Ras-family GTPases bound to GDP to the active form bound to GTP, external signals are transmitted from receptors on the cytoplasmic membrane to the interior of the cell [47]. The adaptor complex then activates Ras-GTP. Post RAS activation, there is a phosphorylation-dependent cascade, activating RAF, MEK, and, finally, ERK. The activation of the ERK/MAPK pathway is reported to also induce the synthesis of cyclin D1, relevant in the progression of cell cycle [48]. Moreover, Raf1 creates a link between the MAPK/ERK pathway and PI3K/AKT, allowing the possibility of correlated feedback between the two distinct metabolic paths, both upregulated in the CRC pathology [44].3.3.2. MAPK/JNK Signaling in CRC
The JNK cascade is used by the transforming growth factor-β (TGFβ) in order to autoregulate its concentration; however, it does not affect the JNK protein expression [49]. The pathway activation induced by TGFβ can act in conjunction with SMADs or in SMAD-independent manner [42][44]. The latter is acting on the MKK4–TGFβ-activated kinase 1 (TAK1) axis [49][50], where TAK1 is activated by the tumor necrosis factor-receptor-associated factor 6 (TRAF6), TGFβR2, and TGFβR1 protein complex [49]. Thereafter, it activates the JNK/p38 pathways [49], culminating in the formation of the ICD, domain of TGFβR1, consequent to ubiquitination and TNF-alpha converting enzyme cleavage [43]. ICD is able to translocate into the nucleus, where it induces the overexpression of Snail, MMP2, and p300 genes [51].3.3.3. MAPK/ p38 Signaling in CRC
The MAPK/p38 pathway becomes activated in response to stressors such as hypoxia, heat shock, and osmotic shock [43]. p38 signaling is required for cell migration and metastasis in both CRC and breast cancer [52][53]. Similar to JNKs, p38 MAPKs are activated through autophosphorylation by MKKs [49][50]. TAK1 and TRAF6 are responsible for the SMAD-independent activation of p38. At this level, there is crosstalk in between TAK1 and the NF-κB-MMP9 pathway, another relevant carcinogenesis pathway reported in CRC. The blockade of p38 MAPK activity leads to the recovery of cell cycle and induction cell death mainly through autophagy [53][54].3.4. p53 Signaling in CRC
TP53, a tumor suppressor gene, is among the most commonly mutated genes in CRC and various other types of cancer, with mutations mainly in the exons 5 to 8 (DNA binding domain) [55][56][57]. p53 is physiologically expressed at low levels, partly due to the negative feedback loops that involve MDM2. MDM2 is a transcriptional target of p53 that mediates the degradation of p53 through negative feedback and by functioning as an E3 ubiquitin-ligase that regulates the ubiquitination of p53 [58][59]. Low levels of p53 expression maintain homoeostasis of the cell cycle and cell death. A homolog of MDM2, namely, MDM4, not regulated by p53, forms heterodimers with MDM2 and can enhance MDM2 induced p53 degradation [58]. As a response to stress factors, such as oncogenes, DNA damage, UV irradiation, free radicals, hypoxia, or deficiencies in nutrients and growth factors, p53 is also activated (Figure 4). Then, it can either repress or transactivate downstream targets that regulate cell cycle arrest, apoptosis, DNA repair, and angiogenesis and metastasis [60]. Upon activation, under normal conditions, it can trigger both the intrinsic, mitochondrial, and the extrinsic, death-receptor-induced, apoptotic pathways [61]. The upregulation of expression takes place for the pro-apoptotic B-cell lymphoma-2 (Bcl-2) proteins, such as Bax, Noxa, and PUMA, while the pro-survival Bcl-2 members are downregulated, under normal conditions. This leads to the permeabilization of the mitochondrial outer membrane, releasing cytochrome-c, which binds to Apaf-1, activating the caspase-9. Thereafter, caspase-9 acts as initiator of the cascade, activating caspase-3, caspase-6, and caspase-7 [62]. Among the p53 upregulated death receptors, the most relevant would be PIDD (p53-induced protein with death domain), DR5 (TRAIL-R2), and Fas (CD95/APO-1), which alongside caspase-8 form the death-inducing signaling complexes acting in a loop and in turn activate p53 [59]. Moreover, the transcription factor (TF), TP53, is additionally involved in the genetic modulation including several miRNAs [55][60]. In the cell cycle, under normal conditions, p53 induces the G1/S and G2/M arrest via interactions with targets such as p21(WAF1), GADD45, retinoblastoma protein (Rb), and 14-3-3σ, also cRRIMA-1). Then, it can either repress or transactivate downstream targets that regulate cell cycle arrest, apoptosis, DNA repair, and angiogenesis and metastasis [121]. Upon activation, under normal conditions, it can trigger both the intrinsic, mitochondrial, and the extrinsic, death-receptor-induced, apoptotic pathways [122]. The upregulation of expression takes place for the pro-apoptotic B-cell lymphoma-2 (Bcl-2) proteins, such as Bax, Noxa, and PUMA, while the pro-survival Bcl-2 members are downregulated, under normal conditions. This leads to the permeabilization of the mitochondrial outer membrane, releasing cytochrome-c, which binds to Apaf-1, activating the caspase-9. Thereafter, caspase-9 acts as initiator of the cascade, activating caspase-3, caspase-6, and caspase-7 [123]. Among the p53 upregulated death receptors, the most relevant would be PIDD (p53-induced protein with death domain), DR5 (TRAIL-R2), and Fas (CD95/APO-1), which alongside caspase-8 form the death-inducing signaling complexes acting in a loop and in turn activate p53 [120]. Moreover, the transcription factor (TF), TP53, is additionally involved in the genetic modulation including several miRNAs [116,121]. In the cell cycle, under normal conditions, p53 induces the G1/S and G2/M arrest via interactions with targets such as p21(WAF1), GADD45, retinoblastoma protein (Rb), and 14-3-3σ, also cRRIMA-1
MET [59].
3.5. NF-κB Signaling in CRC
NF-κB is a heterodimer protein, consisting of the p65 and p50 subunits, which are required for its activation and translocation to the nucleus ( [120].
5.5. NF-κB Signaling in CRC
NF-κB is a heterodimer protein, consisting of the p65 and p50 subunits, which are required for its activation and translocation to the nucleus (
Figure 2) [63][64]. Physiologically, in most quiescent cells it is retained in the cytoplasm by I-kappa B (IκB), which covers its nuclear localization sequence (NLS) [65]. Pathologically, the IκB kinase (IKK) complex, containing the NEMO regulatory subunit and the IKKα and IKKβ catalytic subunits, is upregulated by external stimuli through receptors such as the tumor necrosis factor receptor (TNFR), the Toll-like receptor (TLR), and the T/B cell receptor [63][66]. In turn, it phosphorylates IκB, which then is degraded via the ubiquitin-proteasome pathway, allowing thereafter the nuclear translocation of NF-κB [63]. Inside the nucleus, it triggers down-stream gene expression by binding to the enhancer element of the immunoglobulin kappa light-chain, leading to inflammation and cancer development or progression [66][67][68]. In CRC adenocarcinoma, the abnormal activity of K-RAS is directly proportional with the expression of NF-κB [69].
4) [124,125]. Physiologically, in most quiescent cells it is retained in the cytoplasm by I-kappa B (IκB), which covers its nuclear localization sequence (NLS) [126]. Pathologically, the IκB kinase (IKK) complex, containing the NEMO regulatory subunit and the IKKα and IKKβ catalytic subunits, is upregulated by external stimuli through receptors such as the tumor necrosis factor receptor (TNFR), the Toll-like receptor (TLR), and the T/B cell receptor [124,127]. In turn, it phosphorylates IκB, which then is degraded via the ubiquitin-proteasome pathway, allowing thereafter the nuclear translocation of NF-κB [124]. Inside the nucleus, it triggers down-stream gene expression by binding to the enhancer element of the immunoglobulin kappa light-chain, leading to inflammation and cancer development or progression [127,128,129]. In CRC adenocarcinoma, the abnormal activity of K-RAS is directly proportional with the expression of NF-κB [130].
4. Quercetin Impacts the Growth and Proliferation in CRC
In the development of CRC, cellular growth and proliferation mechanisms need to be altered for the progression of the disease. It is noteworthy that quercetin is documented, in Table 23, as an inhibitor of these processes both in vivo and in vitro [70][71][72][73][74][75][76][77][78][79][80][131,132,133,134,135,136,137,138,139,140,141].
Table 23.
Reported targets of quercetin active in the reduction in cellular growth and proliferation of CRC models alongside their in vivo/in vitro testing system. “↓” arrows are indicating the downregulation, while “?” denotes the lack of specific targets in the respective studies.
5. Quercetin Impacts the Cell Cycle in CRC
Even though highly related to the previously described section, the targets of quercetin in the cell cycle arrest might slightly differ, while the activity is mainly described regarding the phase in which the cells are resting (
Table 3).
4).
Table 34.
Reported targets of quercetin active in cell cycle arrest of CRC models alongside their in vivo/in vitro testing system. “↑” and “↓” arrows are indicating the up- and downregulation, respectively, while “?” denotes the lack of specific targets in the respective studies.
| Cell Cycle Arrest Phase and/or Molecular Targets | Testing System | Reference |
|---|
| At G0/G1 phase |
| In vivo: HCT-116 Xenograft mouse model |
| [ | |||
| 85 | ] | [146] | |
| ? | |||
| At G0/G1 phase | In vitro: HT-29 cell culture | [70] | [131] |
| ? | |||
| At G1 or G2 | In vitro: HCT-116 cell culture | [77] | [138] |
| ? | |||
| At G2/M | In vitro: HT-29, HCT116 and SW480 cell cultures | [86] | [147] |
| ↓ p-AKT | |||
| ↑ Cyclin B1 | |||
| At G2/M | In vitro: RKO cell culture | [87] | [148] |
| ↓ CDK1, CDC25c, Cyclin B1 | |||
| ↑ p21 | |||
| At G2/M | In vitro: SW620 cell culture | [88] | [149] |
| ↑ p21, p58 | |||
| ↓ CDC6, CDK4, Cyclin D1 | In vitro: Caco-2 cell culture | [89] | [151] |
| ↓ Ki67 | In vitro: SW480 cell culture | [90] | [152] |
| ↓ Bcl-2 | In vitro: HT-29 cell culture | [70] | [131] |
| ↑ Bax, p53, Caspase-3 |
Abbreviations: Bax = Bcl-2 Associated X-protein; Bcl-2 = B-cell lymphoma 2; CDC6 = Cell division cycle 6 regulatory protein; CDC25c = Cell division cycle 25c regulatory protein; CDK1 = Cyclin dependent kinase 1; CDK4 = Cyclin dependent kinase 1; Ki67 = nonhistone nuclear protein KI67; p-AKT = phosphorylated Protein kinase B; p21 = Cyclin-dependent kinase inhibitor 1; p53 = Tumor protein p53; p58 = p58 Natural killer cell inhibitory receptor.
6. Quercetin Impacts Apoptosis in CRC
Alongside suppression of proliferation, induction of apoptosis increases the theoretical benefits of an anti-cancer agent. In this regard, quercetin takes both approaches against cancer development, with a plentitude of targets, mainly pertaining to signal transduction pathways (
Table 4).
5).
Table 45.
Reported targets of quercetin active in induction of apoptosis of CRC models alongside their in vivo/in vitro testing system. “↑” and “↓” arrows are indicating the up- and down-regulation, respectively, while “?” denotes the lack of specific targets in the respective studies.
| Molecular Targets | Testing System | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ↑ JNK, c-Jun | In vitro: Caco-2 and DLD-1 cell cultures | [71] | [132] | ||||||
| ↑ COX-2 | In vitro: HT-29 and HCT-15 cell cultures | [72] | [133] | ||||||
| ↑ Caspase-3, Cytochrome-c | |||||||||
| ↑ Bax, PARP, APC | In vivo: Wistar rats | [73] | [134] | ||||||
| ↓ Bcl-2, β-catenin | |||||||||
| ↓ p-ERK, KRAS | In vitro: HCT-15 cell culture | [74] | [135] | ||||||
| ↓ TSC22 domain family 3 | In vivo: F344 rats | [74] | [135] | ||||||
| ↓ p-AKT, KRAS | In vitro: CO115 cell culture | [74] | [135] | ||||||
| ↓ PI3K, AKT, p-AKT, Bcl-2 | In vitro: HCT-116 and HT29 cell cultures | [75] | [136] | ||||||
| ↑ Bax | |||||||||
| ↑ Caspase-3, p-JNK | In vitro: DLD-1 | KRASG13D | and DLD-1 | KRASWT | cell cultures | [76] | [137] | ||
| ↓ p-AKT | |||||||||
| ↑ Caspase-3, Cytochrome-c | In vitro: RKO and CCD841 cell cultures | [79] | [140] | ||||||
| ↓ AMPK, HIF-1 | In vitro: HCT-116 | [85] | [146] | ||||||
| ↓ Bcl-2 | In vitro: RKO cell culture | [87] | [148] | ||||||
| ↑ Bax, cleaved-Caspase-3, cleaved-Caspase-9 | |||||||||
| ↑ Bax, Cytochrome-c, Caspase-9, Apaf-1, Caspase-3 | In vitro: SW620 cell culture | [88] | [149] | ||||||
| ↓ GPx, Catalase | |||||||||
| ↓ PI3K, AKT ↑ Caspase-3, Bax | In vitro: SW480 cell culture | [90] | [152] | ||||||
| ↑ PARP, cleaved-Caspase-3, cleaved-Caspase-9 | In vitro: CT-26 cell culture | [91] | [153] | ||||||
| ↓ Bcl-2, Bcl-xL | |||||||||
| ↓ MMP-2, MMP-9, N-cadherin, β-catenin, Snail | In vivo: mouse model of CRC lung metastasis | [91] | [153] | ||||||
| ↑ E-cadherin | |||||||||
| ↑ p53, BAX, p-p38 ↓ Bcl-2 |
In vitro: HCT-15 cell culture | [92] | [154] | ||||||
| ↑ p53, cleaved-Caspase 3, cleaved-Caspase 9, PARP, cleaved-PARP ↓ Bcl-2 |
In vitro: CO115 cell culture | [92] | [154] | ||||||
| ↑ Bax, Caspase-3, Caspase-9 | In vitro: Caco-2 and SW-620 cell cultures | [93] | [155] | ||||||
| ↓ Bcl-2, NF-κB | |||||||||
| ↓ MMP | In vitro: DLD-1 cell culture | [94] | [156] | ||||||
| ↓ MMP | In vitro: HCT-116 cell culture | [95] | [157] | ||||||
| ↑ SIRT-2, p-AMPK, p-p38 | In vitro: HCT-116 cell culture | [96] | [158] | ||||||
| ↓ p-mTOR | |||||||||
| ↑ Caspase-3, cleaved-PARP, p-p38 | In vitro: DLD-1 cell culture | [97] | [160] | ||||||
| ↓ Bcl-2, Cyclin D1, | In vitro: Colo320 cell culture | [98] | [161] | ||||||
| ↑ Bax, Caspase-3, Wnt1, Catalase | |||||||||
| ? | In vivo: AOM/DSS-treated wild-type C57BL/6J mice | [99] | [162] | ||||||
| ? | In vitro: HCT-116 cell culture | [77] | [138] | ||||||
| ? | In vitro: HCT-116 cell culture | [80] | [141] | ||||||
| ? | In vitro: HCT-116 | p53-wt | , HCT-116 | p53-null | , HCT-15 | KRAS-mutated | cell culture | [92] | [154] |
| ? | In vitro: CT-26 cell culture | [100] | [163] |
| Molecular Targets |
|---|
| Testing System |
|---|
| Reference |
|---|
| ↓ p-AKT, MYC |
| In vitro: HT-29 cell culture |
| ↓ CB1 receptor, Wnt/β-catenin, p-GSK3β, |
| In vitro: Caco-2 and DLD-1 cell cultures |
| p-PI3K, p-AKT, p-S6, p-4E-BP1, p-STAT3 |
| ↓ p-AKT, p-GSK3β, Cyclin D1 |
| In vitro: HT-29 and HCT-15 cell cultures |
| ↓ PCNA |
| In vivo: Wistar rats |
| ↓ ANXA1 |
| In vivo: F344 rats |
| ? |
| In vitro: HCT-116 and HT-29 cell cultures |
| ? |
| In vitro: DLD-1 |
| KRASG13D |
| , DLD-1 |
| KRASWT |
| , SW480 |
| KRASG12V |
| , HCT-116 |
| KRASG13D |
| , Colo205 |
| KRASWT |
| , WIDR |
| KRASWT |
| , and HT-29 |
| KRASWT |
| cell cultures |
| ? |
| In vitro: HCT-116 cell culture |
| ? |
| In vitro: HCT15 and CO115 cell cultures |
| ? |
| In vitro: HCT-116 cell culture |
| ? |
| In vitro: RKO and CCD841 cell cultures |
| ? |
| In vivo: F344 AOM treated rats |
Abbreviations: ANXA1 = Annexin A1; CB1 receptor = Cannabinoid receptor type 1; MYC = Myelocytomatosis oncogene product; p-4E-BP1 = phosphorylated Eukaryotic translation initiation factor 4E binding protein 1; p-AKT = phosphorylated Protein kinase B; p-GSK3β = phosphorylated Glycogen synthase kinase 3 beta; p-PI3K = phosphorylated Phosphoinositide 3-kinase; p-S6 = phosphorylated Ribosomal protein S6; p-STAT3 = phosphorylated Signal transducer and activator of transcription 3; PCNA = Proliferating cell nuclear antigen; Wnt/β-catenin = Wingless-related integration site/β-catenin pathway.
Quercetin inhibits in cell culture the activity of AKT (also known as protein kinase B) by hindering it from phosphorylation, thus decreasing the concentration of p-AKT, in several CRC representative cell lines, such as HT-29 [131,133], Caco-2 [132], DLD-1 [132], and HCT-15 [133].
In vivo, Wistar rats and F344 rats, studies claim a decrease in proliferating cell nuclear antigen (PCNA) and annexin A1 (ANXA1) upon dietary ingestion of quercetin in comparison to the control group [134,135]. PCNA, originally a DNA sliding clamp for replicative polymerases and vital component of the eukaryotic chromosomal DNA replisome, has been revealed to interact with multiple partners, involved in DNA repair, Okazaki fragment processing, DNA methylation, and chromatin remodeling [142]. ANXA1, also known as lipocortin I, is a member of the annexin multigene superfamily of Ca
Abbreviations: AKT = Protein kinase B; AMPK = 5′ adenosine monophosphate-activated protein kinase; Apaf-1 = Apoptotic protease activating factor 1; APC = Adenomatous polyposis coli; Bax = Bcl-2 Associated X-protein; Bcl-2 = B-cell lymphoma 2; Bcl-xL = B-cell lymphoma extra-large; c-Jun = AP-1 transcription factor subunit; COX-2 = cyclooxygenase-2; GPx = Glutathione peroxidase; HIF-1 = Hypoxia-inducible factor 1; JNK = c-Jun N-terminal kinases; KRAS = Kirsten rat sarcoma virus; MMP = Matrix metalloproteinases; MMP-2 = Matrix metalloproteinase 2; MMP-9 = Matrix metalloproteinase 9; NF-κB = Nuclear factor kappa-light-chain-enhancer of activated B-cells; p-AKT = phosphorylated Protein kinase B; p-AMPK = phosphorylated 5′ adenosine monophosphate-activated protein kinase; p-ERK = phosphorylated Extracellular signal-regulated kinase; p-JNK = phosphorylated c-Jun N-terminal kinases; p-mTOR = phosphorylated Mammalian target of rapamycin; p-p38 = phosphorylated Mitogen-activated protein kinase p38; PARP = Poly (ADP-ribose) polymerase; PI3K = Phosphoinositide 3-kinases; SIRT-2 = NAD-dependent deacetylase sirtuin 2; Snail = Zinc finger protein SNAI1; TSC22 domain family 3 = Glucocorticoid-induced leucine zipper protein; Wnt1 = Proto-oncogene Wnt-1.
7. Quercetin Impacts Tumor Size in CRC
In the case of the PI3K/AKT and p53 pathways, the quercetin-induced modulation is in accordance with its expected anticarcinogenic activity [90]. However, in the case of the MAPK cascades, the modulation seems to act in anti-apoptotic and pro-proliferative manner, even though the quantification of the tumor size contradicts this in the testing system [91]. Nevertheless, all studies attest that the tumors decreased in size upon quercetin supplementation, supporting, through an additional argument, the potential benefits of testing quercetin alongside chemo- and radiotherapy in clinical setting.In the case of the PI3K/AKT and p53 pathways, the quercetin-induced modulation is in accordance with its expected anticarcinogenic activity [152]. However, in the case of the MAPK cascades, the modulation seems to act in anti-apoptotic and pro-proliferative manner, even though the quantification of the tumor size contradicts this in the testing system [153]. Nevertheless, all studies attest that the tumors decreased in size upon quercetin supplementation, supporting, through an additional argument, the potential benefits of testing quercetin alongside chemo- and radiotherapy in clinical setting.