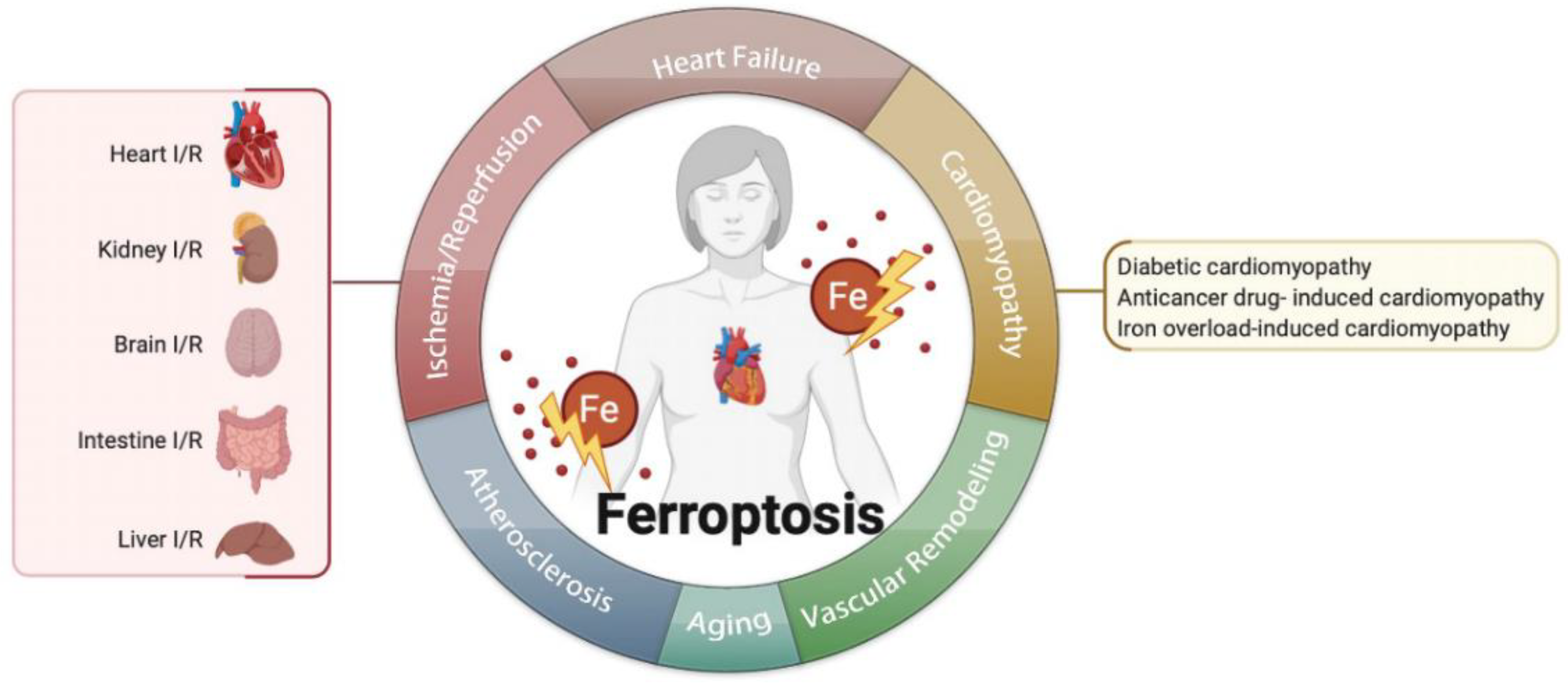
心血管疾病(CVDs)仍然是全球死亡和残疾的主要原因,严重影响人们的生活。由于这些疾病的严重性和复杂性,寻找新的调节机制来治疗心血管疾病非常重要。铁死亡是目前正在研究的一种新型调节性细胞死亡。越来越多的证据表明,铁死亡在心血管疾病中起着重要作用,例如在缺血/再灌注损伤、心力衰竭、心肌病和动脉粥样硬化中。通过靶向铁死亡来预防心血管疾病是一种很有前途的方法;因此,在这篇综述中,我们总结了铁死亡的最新调控机制和目前与每种 CVD 相关的研究,然后对 CVD 的铁死亡治疗以及这一有趣生物学的未来方向提出了批判性观点。Cardiovascular diseases (CVDs) are still a major cause of global mortality and disability, seriously affecting people’s lives. Due to the severity and complexity of these diseases, it is important to find new regulatory mechanisms to treat CVDs. Ferroptosis is a new kind of regulatory cell death currently being investigated. Increasing evidence showed that ferroptosis plays an important role in CVDs, such as in ischemia/reperfusion injury, heart failure, cardiomyopathy, and atherosclerosis.
- ferroptosis
- cardiovascular diseases
- heart failure
- cardiomyopathy
- atherosclerosis
一、简介1. Introduction
2. 铁死亡的调节途径2. The Regulating Pathways of Ferroptosis
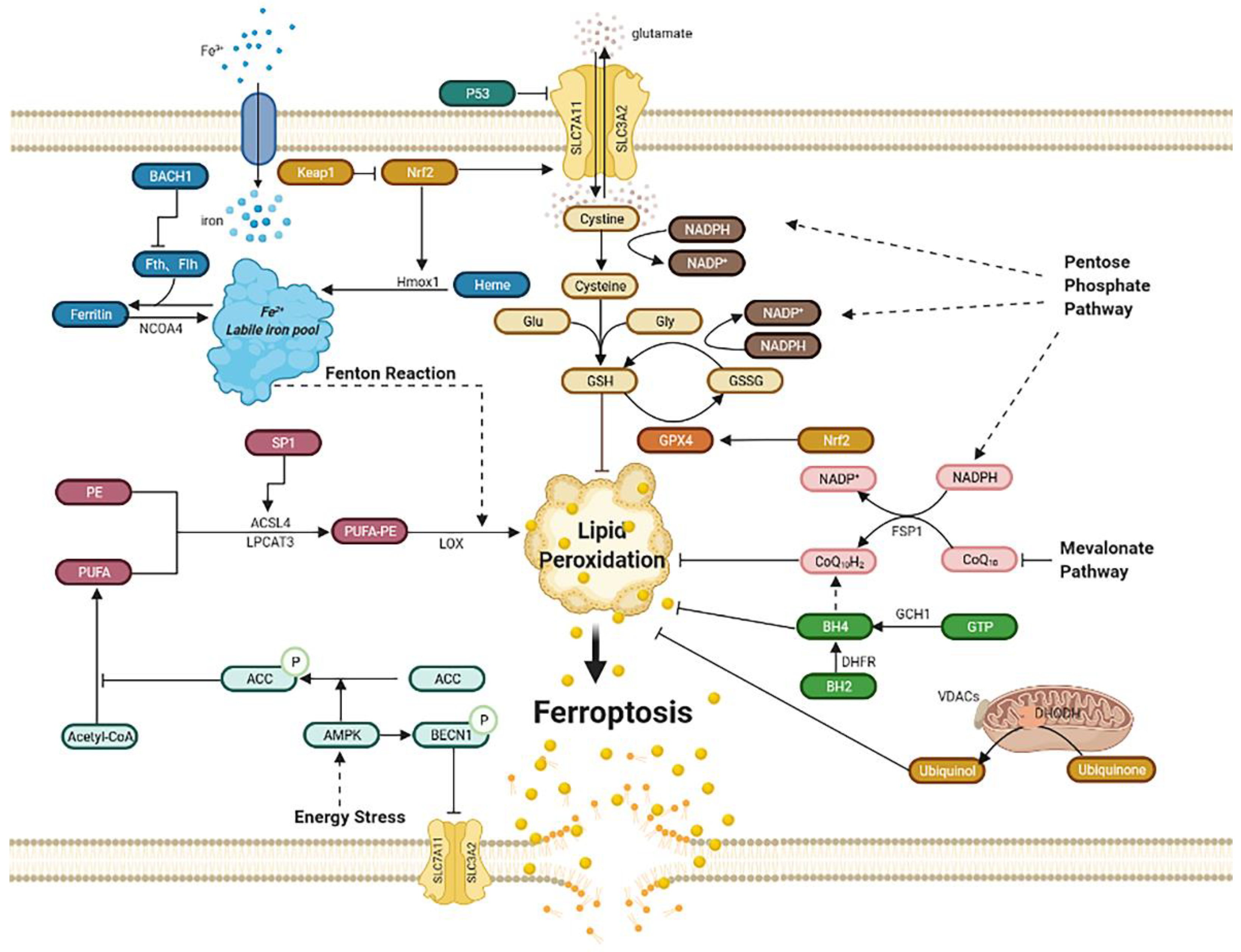
2.1. Iron Metabolism
2.2. Lipid Peroxidation
2.3. Xc-GSH-GPX4 Pathway
2.4. FSP1–CoQ10–NAD(P)H Pathway
2.5. Mitochondria
2.6. GCH1–BH4–DHFR Pathway
2.7. Energy Stress
3. Ferroptosis in Cardiovascular Diseases
3.1. Ferroptosis in Ischemia/Reperfusion (I/R)
| Organs | Models and Cells | Compounds | Targets | Mechanisms and Consequences |
References |
|---|---|---|---|---|---|
| heart | Sprague–Dawley rats, H9c2 cell | Fer-1 | ACSL4; GPX4 | ACSL4 reduced; GPX4 increased; infarct size reduced. | [9] |
| heart | Mice (C57BL/6J) | Fer-1 | AA | 5-HETE, 11-HETE, 12-HETE, and 15-HETE reduced; infarct size reduced; left ventricular improved. | [8] |
| heart | Mice (C57BL/6J) | Lip-1 | GPX4; ROS | GPX4 increased; ROS decreased; infarct size reduced; mitochondrial structural integrity and function maintained. | [10] |
| renal | HK-2 cells | ALR | GPX4; ROS | ROS decreased; ALR and GPX4 colocalized; renal I/R injury protected. | [67][54] |
| renal | Mice | 2DG; AICAR; Fer-1 | AMPK; PUFA | AMPK activation; renal I/R injury protected. | [51] |
| renal | Mice (C57BL/6) HK-2 cells | Panx1 deletion |
Hmox1, NCOA4, Fth1 |
Hmox1 upregulated, NCOA4 and FTH1 inhibited; lipid peroxidation decreased; renal I/R injury protected. | [66][53] |
| renal | Mice (C57BL/6) | Fer-1 | renal I/R injury protected. | [72][59] | |
| intestine | Mice(C57BL/6) Caco-2 cells | Lip-1; ROSI | GPX4 ACSL4 | GPX4 induced; ACSL4 inhibited; intestine I/R injury protected. | [34] |
| brain | Mice (C57BL/6) PC12 cells | LV-shRNA-PVT1 or LV-miR-214 | GPX4, SLC7A11 | GPX4 and SLC7A11 increased; infarct size reduced. | [69][56] |
| brain | Mice (C57BL/6) | Lip-1; Fer-1 | infarct size reduced. | [73][60] | |
| brain | Mice (Sv129/J) purified cortical neurons. | Desferrioxamine | HIF-1 | HIF-1 increased; tolerance against reversible focal cerebral ischemia. | [74][61] |
| liver | Mice (C57BL/6) | Lip-1 | liver I/R injury reduced. | [70][57] |
3.2. Ferroptosis in Heart Failure
3.3. Ferroptosis in Cardiomyopathies
3.3.1. Anticancer Drug-Induced Cardiomyopathy
3.3.2. Diabetic Cardiomyopathy
3.3.3. Iron Overload-Induced Cardiomyopathy
3.4. Ferroptosis in Atherosclerosis
3.5. Ferroptosis in Aging
3.6. Ferroptosis in Vascular or Ventricular Remodeling
References
- Xiao, F.J.; Zhang, D.; Wu, Y.; Jia, Q.H.; Zhang, L.; Li, Y.X.; Yang, Y.F.; Wang, H.; Wu, C.T.; Wang, L.S. miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem. Biophys. Res. Commun. 2019, 515, 448–454.
- Wang, Y.; Tang, M. PM2.5 induces ferroptosis in human endothelial cells through iron overload and redox imbalance. Environ. Pollut. 2019, 254, 112937.
- Sampilvanjil, A.; Karasawa, T.; Yamada, N.; Komada, T.; Higashi, T.; Baatarjav, C.; Watanabe, S.; Kamata, R.; Ohno, N.; Takahashi, M. Cigarette smoke extract induces ferroptosis in vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H508–H518.
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290.
- Li, Z.; Lin, L.; Wu, H.; Yan, L.; Wang, H.; Yang, H.; Li, H. Global, Regional, and National Death, and Disability-Adjusted Life-Years (DALYs) for Cardiovascular Disease in 2017 and Trends and Risk Analysis From 1990 to 2017 Using the Global Burden of Disease Study and Implications for Prevention. Front. Public Health 2021, 9, 559751.
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25.
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021.
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304.
- Li, W.; Li, W.; Leng, Y.; Xiong, Y.; Xia, Z. Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury through Endoplasmic Reticulum Stress. DNA Cell Biol. 2020, 39, 210–225.
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611.
- Dabkowski, E.R.; Williamson, C.L.; Hollander, J.M. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Rad. Biol. Med. 2008, 45, 855–865.
- Chen, X.; Xu, S.; Zhao, C.; Liu, B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 2019, 516, 37–43.
- Liu, B.; Zhao, C.; Li, H.; Chen, X.; Ding, Y.; Xu, S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem. Biophys. Res. Commun. 2018, 497, 233–240.
- Conrad, M.; Proneth, B. Broken hearts: Iron overload, ferroptosis and cardiomyopathy. Cell Res. 2019, 29, 263–264.
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680.
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Rad. Biol. Med. 2020, 160, 92–102.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Agmon, E.; Solon, J.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 8, 5155.
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849.
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e421.
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698.
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692.
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53.
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360.
- Garcia-Bermudez, J.; Birsoy, K. A mitochondrial gatekeeper that helps cells escape death by ferroptosis. Nature 2021, 593, 514–515.
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841.
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428.
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032.
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell Physiol. 2018, 233, 9179–9190.
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137.
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J. Biol. Chem. 2020, 295, 69–82.
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343.
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Diff. 2019, 26, 2284–2299.
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Sign. Transduct. Target. Ther. 2020, 5, 108.
- Liu, X.; Zhang, Y.; Zhuang, L.; Olszewski, K.; Gan, B. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edged sword in cellular redox regulation. Genes Dis. 2021, 8, 731–745.
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Diff. 2016, 23, 369–379.
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62.
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575.
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front Neurosci. 2020, 14, 267.
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371.
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; DeNicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189.e177.
- Yang, Y.; Chen, J.; Gao, Q.; Shan, X.; Wang, J.; Lv, Z. Study on the attenuated effect of Ginkgolide B on ferroptosis in high fat diet induced nonalcoholic fatty liver disease. Toxicology 2020, 445, 152599.
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937.
- Dai, E.; Zhang, W.; Cong, D.; Kang, R.; Wang, J.; Tang, D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem. Biophys. Res. Commun. 2020, 523, 966–971.
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058.
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 2018, 148, 155–162.
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505.
- Shin, D.; Lee, J.; You, J.H.; Kim, D.; Roh, J.L. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020, 30, 101418.
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201.
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234.
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X(c)(-) Activity. Curr. Biol. 2018, 28, 2388–2399.e2385.
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J. Biol. Chem. 2019, 294, 19395–19404.
- Huang, L.L.; Liao, X.H.; Sun, H.; Jiang, X.; Liu, Q.; Zhang, L. Augmenter of liver regeneration protects the kidney from ischaemia-reperfusion injury in ferroptosis. J. Cell Mol. Med. 2019, 23, 4153–4164.
- Choi, N.; Whitlock, R.; Klassen, J.; Zappitelli, M.; Arora, R.C.; Rigatto, C.; Ho, J. Early intraoperative iron-binding proteins are associated with acute kidney injury after cardiac surgery. J. Thorac Cardiovasc. Surg. 2019, 157, 287–297.e282.
- Lu, J.; Xu, F.; Lu, H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020, 260, 118305.
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191.
- Yan, H.F.; Tuo, Q.Z.; Yin, Q.Z.; Lei, P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool Res. 2020, 41, 220–230.
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841.
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530.
- Prass, K.; Ruscher, K.; Karsch, M.; Isaev, N.; Megow, D.; Priller, J.; Scharff, A.; Dirnagl, U.; Meisel, A. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J. Cereb. Blood Flow Metab. 2002, 22, 520–525.
- Wang, C.; Zhu, L.; Yuan, W.; Sun, L.; Xia, Z.; Zhang, Z.; Yao, W. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J. Cell. Mol. Med. 2020, 24, 6670–6679.
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308.
- Wang, J.; Deng, B.; Liu, Q.; Huang, Y.; Chen, W.; Li, J.; Zhou, Z.; Zhang, L.; Liang, B.; He, J.; et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 2020, 11, 574.
- Ferreira, C.; Bucchini, D.; Martin, M.E.; Levi, S.; Arosio, P.; Grandchamp, B.; Beaumont, C. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 2000, 275, 3021–3024.
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545.
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501.
- Shizukuda, Y.; Matoba, S.; Mian, O.Y.; Nguyen, T.; Hwang, P.M. Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol. Cell Biochem. 2005, 273, 25–32.
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Cardiac Specific Knockout of p53 Decreases ER Stress-Induced Mitochondrial Damage. Front. Cardiovasc. Med. 2019, 6, 10.
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747.
- Ge, Z.D.; Lian, Q.; Mao, X.; Xia, Z. Current Status and Challenges of NRF2 as a Potential Therapeutic Target for Diabetic Cardiomyopathy. Int. Heart J. 2019, 60, 512–520.
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front Neurosci. 2018, 12, 466.
- Wang, Y.; Yu, R.; Wu, L.; Yang, G. Hydrogen sulfide guards myoblasts from ferroptosis by inhibiting ALOX12 acetylation. Cell Sign. 2021, 78, 109870.
- Kremastinos, D.T.; Farmakis, D. Iron overload cardiomyopathy in clinical practice. Circulation 2011, 124, 2253–2263.
- Berdoukas, V.; Coates, T.D.; Cabantchik, Z.I. Iron and oxidative stress in cardiomyopathy in thalassemia. Free Rad. Biol. Med. 2015, 88, 3–9.
- Ghanavat, M.; Haybar, H.; Pezeshki, S.M.S.; Shahjahani, M.; Jodat, H.; Elyasi, M.; Saki, N. Cardiomyopathy in Thalassemia: Quick Review from Cellular Aspects to Diagnosis and Current Treatments. Lab. Med. 2020, 51, 143–150.
- Mattera, R.; Stone, G.P.; Bahhur, N.; Kuryshev, Y.A. Increased release of arachidonic acid and eicosanoids in iron-overloaded cardiomyocytes. Circulation 2001, 103, 2395–2401.
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795.
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695.
- Sullivan, J.L. Iron in arterial plaque: Modifiable risk factor for atherosclerosis. Biochim. Biophys. Acta 2009, 1790, 718–723.
- Xu, S. Iron and Atherosclerosis: The Link Revisited. Trends Mol. Med. 2019, 25, 659–661.
- Hu, X.; Cai, X.; Ma, R.; Fu, W.; Zhang, C.; Du, X. Iron-load exacerbates the severity of atherosclerosis via inducing inflammation and enhancing the glycolysis in macrophages. J. Cell Physiol. 2019, 234, 18792–18800.
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front Pharmacol. 2019, 10, 306.
- Toyokuni, S.; Yanatori, I.; Kong, Y.; Zheng, H.; Motooka, Y.; Jiang, L. Ferroptosis at the crossroads of infection, aging and cancer. Cancer Sci. 2020, 111, 2665–2671.
- Lapenna, D.; Ciofani, G.; Pierdomenico, S.D.; Giamberardino, M.A.; Porreca, E. Iron status and oxidative stress in the aged rabbit heart. J. Mol Cell Cardiol. 2018, 114, 328–333.
- Cotroneo, E.; Ashek, A.; Wang, L.; Wharton, J.; Dubois, O.; Bozorgi, S.; Busbridge, M.; Alavian, K.N.; Wilkins, M.R.; Zhao, L. Iron homeostasis and pulmonary hypertension: Iron deficiency leads to pulmonary vascular remodeling in the rat. Circ. Res. 2015, 116, 1680–1690.
- Naito, Y.; Hosokawa, M.; Sawada, H.; Oboshi, M.; Iwasaku, T.; Okuhara, Y.; Eguchi, A.; Nishimura, K.; Soyama, Y.; Hirotani, S.; et al. Iron is associated with the development of hypoxia-induced pulmonary vascular remodeling in mice. Heart Vess. 2016, 31, 2074–2079.
- Ryals, B.; Westbrook, E.; Schacht, J. Morphological evidence of ototoxicity of the iron chelator deferoxamine. Hear Res. 1997, 112, 44–48.