Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Jessie Wu and Version 3 by Jessie Wu.
Ischemic stroke causes a heavy health burden worldwide, with over 10 million new cases every year. Despite the high prevalence and mortality rate of ischemic stroke, the underlying molecular mechanisms for the common etiological factors of ischemic stroke and ischemic stroke itself remain unclear, which results in insufficient preventive strategies and ineffective treatments for this devastating disease. The transient receptor potential cation channel, subfamily M, member 2 (TRPM2), a non-selective ion channel activated by oxidative stress, is actively involved in all the important steps in the etiology and pathology of ischemic stroke.
- ischemic stroke
- TRPM2
- oxidative stress
1. Transient Receptor Potential Melastatin 2
Transient receptor potential melastatin 2 (TRPM2) is a Ca2+-permeable and non-selective ion channel [1][2][3] belonging to the TRP channel family [4][5][6]. This 172 kDa membrane protein is located in an ~90 kb area on chromosome 21q22.3 in humans [7]. Compared with other members of the TRPM family, TRPM2 shares the characteristic N-terminal homology regions (MHR1–MHR4) and C-terminal coiled-coil domains (CTD), but is featured by its unique C-terminal NUDT9-H domain [8]. The NUDT9-H domain is homologous to NUDT9, a highly conserved adenosine diphosphate ribose (ADPR) pyrophosphatase [8][9]. Therefore, TRPM2 can be referred as a chanzyme. The enzyme function of NUDT9-H varies depending on the species [10][11]. Although invertebrate NUDT9-H is an active ADPRase, vertebrate NUDT9-H does not have the ADPRase activity. However, the NUDT9-H domain is found to be critical for the surface expression of TRPM2 and the gating of TRPM2 by ADPR [12][13]. Besides the cell membrane, TRPM2 is also suggested to be localized in the membrane of lysosomes [14][15].
Upon activation, TRPM2 currents display a characteristic linear I–V relationship, with a single channel conductance about 75–78 pS [16]. TRPM2 is gated by ADPR and Ca2+, and can be inhibited by N-(p-amylcinnamoyl) anthranilic acid (ACA) [17] and 2-aminoethoxydiphenyl borate (2-APB) [17]. Recent atomic structural analysis has revealed a Ca2+ binding site [18][19][20] which plays a major role in channel gating for several TRPM channels including TRPM2, TRPM4, and TRPM8 [21]. Activation of TRPM2 also requires ADPR binding to the channels in the presence of Ca2+. It was well known that ADPR binds to the C-terminal NUDT9-H domain of TRPM2 [22]. Interestingly, recent TRPM2 structures reveal that in zebrafish (Danio rerio) TRPM2 (drTRPM2), besides the NUDT9-H domain, there is also another ADPR binding site at the N terminal MHR1/2 domain [23]. However, this newly discovered binding site in drTRPM2 is not involved in the ADPR gating in human TRPM2 [23], although the antagonist 8-Br-cADPR binds to the MHR1/2 domain. Like many other TRP channels, TRPM2 is a temperature sensor [24]. TRPM2 is activated by heat, and the temperature threshold of TRPM2 is 47.2 ± 0.2 °C, but this threshold can be significantly reduced to a physiologically reachable level (36.3 ± 0.6 °C) by oxidative stress [25][26]. Moreover, oxidative stress also indirectly activates TRPM2 by increasing the production of ADPR and Ca2+ [27]. Therefore, TRPM2 is regarded as a cellular sensor for oxidative stress [28]. The local temperature in the affected tissue during inflammation usually increases [29], and oxidative-stress-mediated Ca2+ signaling is critical for the elicitation of inflammatory responses in immune cells [30]. The combined sensing of heat and oxidative stress confers TRPM2 with a crucial function in regulating inflammatory responses (Figure 1 and Figure 2).
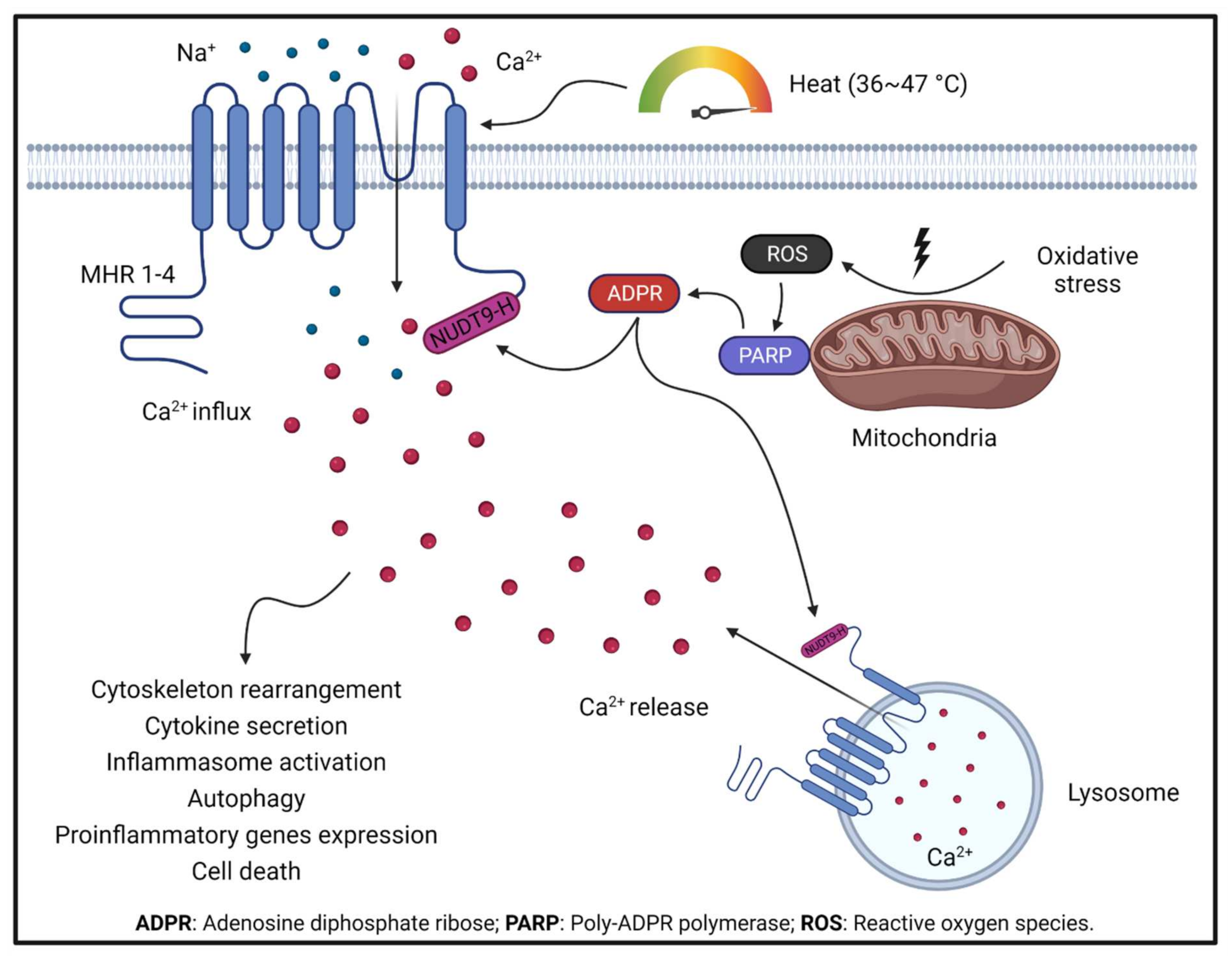
Figure 1. TRPM2 activation by oxidative stress and TRPM2-mediated Ca2+ signaling under oxidative stress boosts ROS production in mitochondria. Increased ROS production activates PARP, which produces ADPR, a potent endogenous TRPM2 activator. ADPR activates TRPM2 by binding to the NUDT9-H domain at the C terminus. TRPM2 activation leads to Ca2+ influx from the extracellular environment and Ca2+ release from lysosomes. TRPM2-mediated Ca2+ signaling is critical in regulating a series of cellular functions.
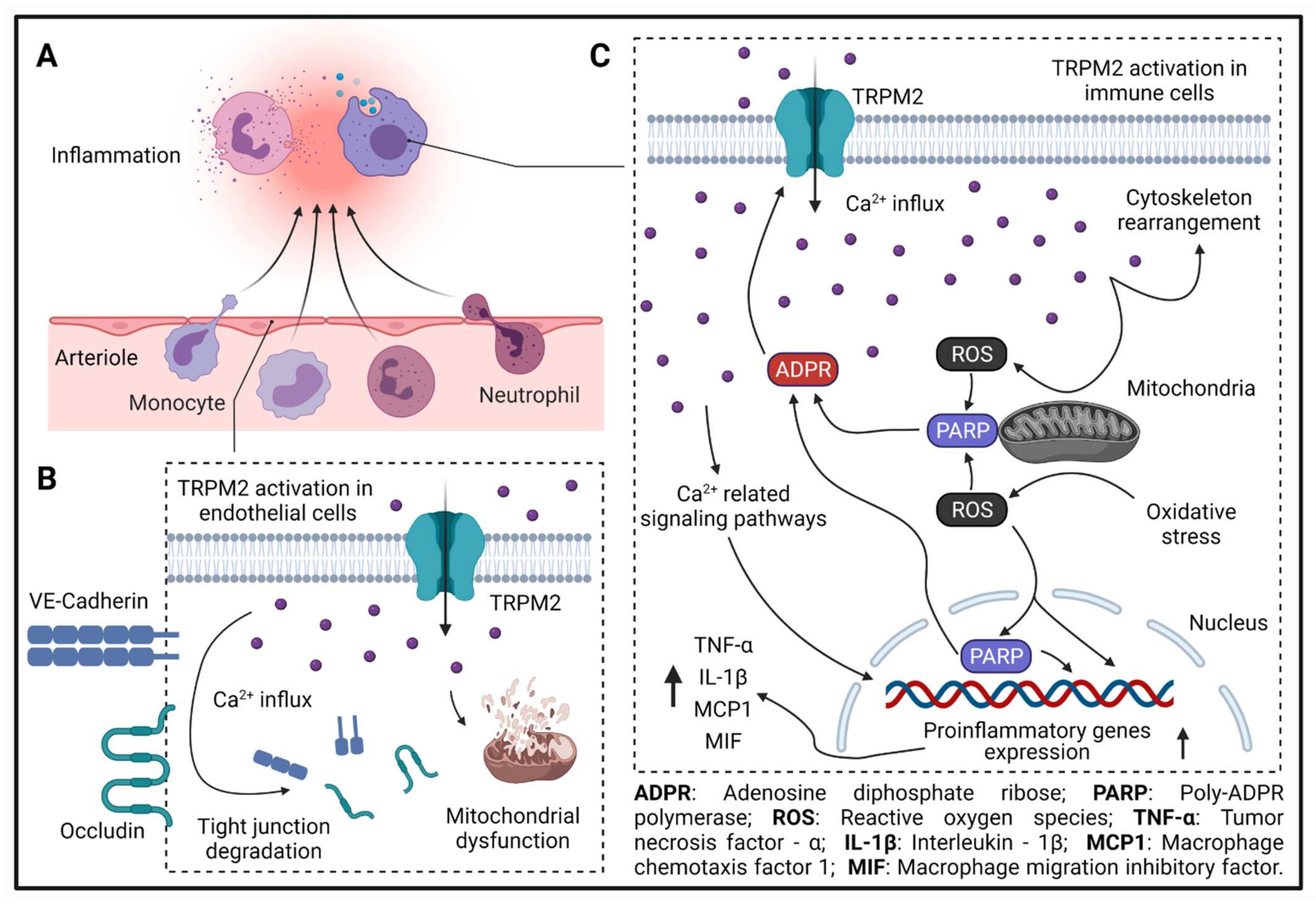
Figure 2. TRPM2 in inflammation. (A) Leukocyte extravasation during inflammation. (B) TRPM2-mediated Ca2+ influx leads to tight-junction molecule degradation (VE-cadherin and occludin) and mitochondrial dysfunction in endothelial cells. (C) TRPM2-mediated Ca2+ influx is needed for immune cell migration and activation. During inflammation, ROS production in mitochondria is increased, which activates PARP in mitochondria or in the nucleus and enhances the production of ADPR. Increased ADPR potentiates TRPM2-mediated Ca2+ influx, which further increases the production of ROS in mitochondria, leading to the formation of a feed-forward vicious cycle. ROS-, PARP-, and Ca2+ -related signaling pathways increase the expression of proinflammatory genes, such as TNF-α, IL-1β, MCP1, and MIF. Moreover, TRPM2-mediated Ca2+ influx promotes cytoskeleton rearrangement and immune cell migration.
TRPM2 is ubiquitously expressed in almost all tissues and cell types [31], and TRPM2-mediated Ca2+ signaling is involved in various important cellular functions, including cytokine/hormone secretion [14][32], cytoskeletal rearrangement [15], cell migration [33], regulation of reactive oxygen species (ROS) production [34], autophagy [35], inflammasome activation [32], and cell death [36]. Therefore, TRPM2 is closely related to many human diseases, such as myocardial infarction [37], ischemic stroke [38][39][40], Alzheimer’s disease [41][42], cardiomyopathy [43], atrial fibrillation [44], hypertension [45], atherosclerosis [46], inflammatory lung injury [34][47], diabetes [48], ischemic kidney disease [49], and many cancers [50]. Besides ischemic stroke itself, almost all the above diseases are the upstream etiological factors for ischemic stroke [51], highlighting the critical and comprehensive role of TRPM2 in the development and progression of this devastating disease.
2. Transient Receptor Potential Melastatin 2 in Diseases Increasing Risk for Ischemic Stroke
The TOAST classification denotes the causes of ischemic stroke into five subtypes: large-artery atherosclerosis, cardio-embolism, small-vessel occlusion, other determined etiology, and undetermined etiology, which is widely accepted by clinicians [52]. Among all the etiological factors of ischemic stroke, the most common ones are atrial fibrillation, hypertension, atherosclerosis, diabetes, and thrombosis [53][54]. Well control of these diseases is important for the prevention of ischemic stroke. However, the molecular mechanisms involved in the development of these diseases are still not completely understood.
2.1. Atrial Fibrillation
Atrial fibrillation (AF) is a common disease in the elderly, and atrium-derived thrombus caused by AF is one of the most common causes of ischemic stroke [55]. Atrial remodeling is the cellular mechanism promoting the development and maintenance of AF [56], in which atrial fibroblasts play a predominant role by increasing the production of extracellular matrix proteins, thereby causing atrial fibrosis [57].
The principal risk factor for AF is aging [58]. Aging is usually closely related to chronic systemic inflammation, which is referred as inflammaging [59]. Chronic inflammatory response is an critical driving force in the development of atrial remodeling by enhancing the fibrotic activity of atrial fibroblasts [60]. TRPM2 is an important regulator of inflammation. Previously, TRPM2 was found to be associated with age-associated inflammatory responses in the brain, and deletion of TRPM2 protected mice against age-associated cognitive function caused by inflammation [61]. Moreover, loss of glutathione, a physiological antioxidant, during neuron senescence facilitates TRPM2 activation [62]. Aging-related chronic inflammation is also critical in the development of many cardiovascular diseases [59], and previously resesachers found that the expression of TRPM2 was significantly increased in atrial fibroblasts isolated from patients with AF compared with that from non-AF patients [63]. Therefore, TRPM2 might also play an important role in the aging-related chronic inflammation in the atria, thereby promoting the development of atrial remodeling and AF.
Oxidative stress promotes AF development by impairing the contractility of atrial myocytes [64][65] and accelerating atrial fibrosis [66]. In a recent paper we showed that TRPM2 activation markedly and rapidly promoted the production of ROS in mitochondria of macrophages [46]. TRPM2 itself is a cellular sensor for oxidative stress [28]. Therefore, the increased production of ROS will in turn further promote the activation of TRPM2, thereby forming a feed-forward vicious cycle [46]. Moreover, Ca2+ signaling is critical for the activation of fibroblasts during atrial fibrosis [67][68], and TRPM2-mediated Ca2+ influx under oxidative stress has been shown to be required for many cellular functions [14][32][33][35]. Considering the high expression of TRPM2 in atrial tissue after AF [63], there is a high possibility that TRPM2 also contributes to the progression of AF by magnifying the oxidative stress response and Ca2+ signaling in atrial myocytes and fibroblasts.
Intracellular Ca2+ is not only critical for regulating the mechanical and electrical activity of healthy atrial muscle, but it also plays an important role in the triggering of AF [69]. AF can be triggered by afterdepolarizations [69]. There are two types of afterdepolarizations, early afterdepolarizations (EAD) and delayed afterdepolarizations (DAD). Both EAD and DAD can cause abnormal electrical activities in atrial muscle, and Ca2+ overload during EAD in atrial muscle was shown to trigger AF [70]. The molecular mechanisms of EAD remain mysterious. Some studies suggest that EAD might result from the Ca2+ influx from L-type Ca2+ channels or spontaneous Ca2+ release from the endoplasmic reticulum (ER) [71]. Considering the important role and active involvement of aging and oxidative stress in the development of AF and TRPM2 in these two conditions, TRPM2-mediated Ca2+ influx or Ca2+ release from lysosome might also contribute to the Ca2+ overload during EAD and the triggering of AF.
2.2. TRPM2 in Hypertension
Hypertension, perhaps the most prevalent disease in humans, is an independent risk factor for ischemic stroke [72]. Traditionally, hypertension was thought to cause stroke mainly in the elderly and young males [73][74]. Recently, a small increase in blood pressure even as mild as 10 mm Hg was shown to be associated with a 38% increased risk of stroke in females [75]. The characteristic pathological change of hypertension is arteriosclerosis, which increases the peripheral resistance and blood pressure [76]. The progressive hardening of arteriole walls is called arterial remodeling [77], in which endothelial dysfunction and smooth-muscle proliferation play a central role [76].
Oxidative stress has long been known to promote the development and progression of hypertension by inducing endothelial dysfunction [78], smooth muscle hypertrophy [79], and vascular remodeling [80], whereas antioxidants were shown to have a protective effect against hypertension by preventing the vascular dysfunction [81]. However, the underlying molecular mechanisms remain unknown [82]. In a recent study, the Ca2+ dysregulation and hyperactivity of vascular smooth muscle cells during hypertension was found to be dependent on TRPM2-mediated Ca2+ influx, which is activated by angiotensin-II-mediated increase of ROS production [45]. Similarly, endothelial dysfunction mediated by ROS-activated TRPM2 was also found to accelerate the development of Alzheimer’s disease [41] and aggregate inflammatory lung injury [47]. As a cellular sensor for oxidative stress, TRPM2 might mediate the detrimental effects of ROS on endothelial cells and smooth muscle cells in the development of hypertension.
Intracellular Ca2+ is a critical regulator of endothelial function [83] and smooth muscle contractility [84]. Ca2+ signaling regulates the expression of endothelial nitric oxide synthase [83][85], whereas blockade of Ca2+ channels enhanced the production of nitric oxide, a potent vasodilator, in endothelial cells [86][87]. Moreover, increase of intracellular Ca2+ in smooth muscle cells enhanced muscle tone and peripheral resistance [88]. In platelets isolated from hypertensive patients, cellular Ca2+ concentration was much higher than that from patients with normal blood pressure [89]. There are two sources of cytosolic free Ca2+—one is the Ca2+ influx from the extracellular environment, and another is the Ca2+ release from intracellular organelles, such as the endoplasmic reticulum and lysosomes. The multiple roles of TRPM2-mediated Ca2+ influx have been well documented in many cell types including endothelial cells [47]. However, there are a limited number of studies reporting the function of TRPM2 in lysosomes. The lysosome is the most important organelle for autophagy and TRPM2 has long been shown to be associated with autophagy [90]. Lysosomal TRPM2-mediated Ca2+ release was shown to be responsible for the pancreatic β-cell death under oxidative stress [14]. Recently TRPM2-mediated Ca2+ release from lysosomes was found to promote autophagic degradation in vascular smooth muscle cells, thereby causing cell death [91], and knockout of TRPM2 attenuated hypertension in spontaneously hypertensive rats by reconstituting autophagy in endothelial cells and vascular smooth muscle cells [92]. In summary, TRPM2-mediated Ca2+ signaling aggregates the dysfunction of endothelial cells and vascular smooth muscle cells in the development and progression of hypertension.
2.3. TRPM2 in Atherosclerosis
Atherosclerosis is a dangerous risk factor for ischemic stroke. Atherosclerotic plaque on the aortic arch [93] and carotid artery [94] significantly increases the risk of ischemic stroke. Moreover, break of atherosclerotic plaque in cerebral arteries directly leads to the formation of in situ thrombus [95], which is usually more difficult to evaluate and predict due to the small plaque size and deep position in the skull [95]. The central pathological feature of atherosclerosis is foam cell formation [96]. Uptake of too much cholesterol transforms infiltrated macrophages into highly inflammatory foam cells, which are the culprit in the development and progression of atherosclerotic plaque [97]. Foam-cell formation includes two critical processes—one is macrophage infiltration, the another is phagocytosis of cholesterol included in oxidized low-density-lipoprotein (oxLDL) [97].
The first step of macrophage infiltration during atherosclerosis is macrophage chemotaxis toward the lesion site, which is caused by chemokines. Previously, TRPM2 was found to be critical for the chemotaxis of neutrophils by formyl-methionyl-leucyl-phenylalanine (fMLP) [98]. fMLP is also a well-established chemokine for macrophages, suggesting the potential role of TRPM2 in regulating macrophage chemotaxis. Indeed, in a recent study, we found that the in vitro macrophage migration induced by macrophage chemotaxis protein 1 (MCP1) was inhibited by deleting TRPM2 or inhibiting the activation of TRPM2 [46]. MCP1 secreted by endothelial cells in response to subendothelial deposition of oxLDL is one of the initial driving forces of macrophage infiltration during atherosclerosis [97]. Similar to our findings, hHydrogen peroxide (H2O2) was shown to attract neutrophil migration both in vivo and in intro, which was also abolished by TRPM2 knockout or TRPM2 inhibition [99]. H2O2 is an important molecular signal generated during inflammation [100]. H2O2 gradient produced by wounded tissue is required for the rapid leukocyte recruitment after injury [101]. Like the chemotactic effect on neutrophils, H2O2 might also be an important chemokine for macrophages, and the recruitment of macrophages mediated by H2O2 might depend on TRPM2 activation.
The second step of macrophage infiltration into the vessel wall is attachment and penetration of the endothelium, which is called extravasation [97]. Leukocyte extravasation is mediated by several surface-adhesion molecules. TRPM2 was found to be required for the transendothelial migration of neutrophils induced by endotoxin, in which TRPM2-mediated Ca2+ influx promotes the phosphorylation of VE-cadherin and degradation of tight junctions between endothelial cells [33]. Similarly, specific deletion of TRPM2 in immune cells markedly decreased immune-cell invasion into the brain after ischemic stroke [38]. Moreover, in mice fed with high-fat diet, Trpm2 deletion caused reduced macrophage infiltration and attenuated inflammation in adipose tissue compared with wild type mice [102]. In our study, we also found that TRPM2-mediated Ca2+ influx is also needed for the in vitro transendothelial migration of macrophages induced by MCP1, and Trpm2 deletion reduces the macrophage burden in atherosclerotic plaques in vivo [46]. These studies highlight the crucial role of TRPM2 in promoting macrophage infiltration during atherogenesis.
After infiltrating into the vessel wall, macrophages engulf oxLDL and become highly proinflammatory foam cells [97]. Foam cells promote the development and progression of atherosclerosis by secreting pro-inflammatory cytokines, chemokines, and tissue-degrading enzymes, which cause profound inflammatory responses and lead to lesion expansion [96]. Activation of the NFκB signaling pathway is required for the activation of macrophages, and TRPM2-mediated Ca2+ signaling has been shown to be indispensable for NFκB signaling activation in macrophages during inflammation, suggesting the potential tole of TRPM2 in transforming macrophages into foam cells [103]. CD36 is the most important receptor for oxLDL uptake in macrophages, and activation of the CD36 downstream signaling pathways is required for macrophage activation and subsequent foam-cell formation [104][105][106][107][108][109]. Previously TRPM2 was shown to mediate the activation of macrophages during infection [110][111] and inflammation [32][112] or when temperature increases [25]. We found that TRPM2 is required for CD36 activation and oxLDL uptake in macrophages, and activation of CD36 by oxLDL further promotes the activation of TRPM2, thereby forming a feed-forward viscous cycle [46]. NLRP3 inflammasome activation by engulfed cholesterol is required for macrophage activation during atherosclerosis [113]. We also found that NLRP3 inflammasome activation by oxLDL is dependent on TRPM2-mediated Ca2+ signaling [46]. All these studies suggest TRPM2 is likely to play a crucial role in the transformation of infiltrated native macrophages into pro-inflammatory foam cells.
2.4. TRPM2 in Diabetes
Hyperglycemia causes a series of pathological changes in the vessel walls, including endothelial dysfunction, basal membrane thickening, interstitial fibrosis, and vessel stiffness, which markedly increase the risk of developing ischemic stroke [114]. Moreover, the mortality of ischemic stroke is higher and clinical outcomes are poorer in patients with diabetes [115]. Well-controlled hyperglycemia significantly decreases the risk of ischemic stroke, decreases mortality, and improves clinical outcomes [116]. The prominent pathological feature of diabetes is insufficient insulin secretion (type 1 diabetes mellitus, T1DM) or unresponsiveness of peripheral tissues to insulin (type 2 diabetes mellitus, T2DM). Many studies have demonstrated that TRPM2 is implicated in the development of both T1DM and T2DM.
T1DM is featured by gradual loss of pancreatic β cells by chronic inflammation in pancreatic islets, thereby resulting in lack of insulin secretion [117]. Ca2+ influx is required for insulin secretion [118], which is further amplified by intracellular Ca2+ release [119]. TRPM2 has a high expression level in pancreatic β cells and activation of TRPM2-mediated Ca2+ influx by ADPR is required for insulin secretion, which can be further enhanced when temperature increases [120]. Knockout of TRPM2 leads to decreased serum insulin levels, increased glucose levels in plasma, and higher insulin sensitivity of peripheral tissues [48][102]. However, excessive Ca2+ influx also leads to cell death under pathological conditions. Both TRPM2-mediated Ca2+ influx and lysosomal Ca2+ release were found to be involved in β-cell death caused by H2O2 [14][121]. Autoimmune inflammatory responses play a central role in the destruction of β cell during T1DM [117]. During chronic inflammation, infiltrated self-reactive T cells and macrophages can produce substantial amount of H2O2, and oxidative stress is also an important mechanism for β-cell dysfunction during T1DM [117][122][123][124]. Moreover, infiltrated immune cells also secrete cytokines such as tumor necrosis factor-α (TNF-α) [125], and TRPM2-mediated Ca2+ influx has been shown to mediated the cytotoxic effect of TNF-α [3][14]. Therefore, TRPM2 is a key molecule in promoting β-cell death in the development of T1DM.
References
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Yokoi, H.; Matsushime, H.; Furuichi, K. Immunocyte Ca2+ Influx System Mediated by LTRPC2. Science 2001, 293, 1327–1330.
- Perraud, A.-L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599.
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173.
- Montell, C.; Birnbaumer, L.; Flockerzi, V. The TRP Channels, a Remarkably Functional Family. Cell 2002, 108, 595–598.
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524.
- Nilius, B.; Voets, T.; Peters, J. TRP Channels in Disease. Sci. STKE 2005, 1–9, .
- McQuillin, A.; Bass, N.J.; Kalsi, G.; Lawrence, J.; Puri, T.; Choudhury, K.; Detera-Wadleigh, S.D.; Curtis, D.; Gurling, H.M.D. Fine mapping of a susceptibility locus for bipolar and genetically related unipolar affective disorders, to a region containing the C21ORF29 and TRPM2 genes on chromosome 21q22.3. Mol. Psychiatry 2005, 11, 134–142.
- Huang, Y.; Winkler, P.A.; Sun, W.; Lü, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 2018, 562, 145–149.
- Perraud, A.-L.; Shen, B.; Dunn, C.A.; Rippe, K.; Smith, M.K.; Bessman, M.J.; Stoddard, B.L.; Scharenberg, A.M. NUDT9, a Member of the Nudix Hydrolase Family, Is an Evolutionarily Conserved Mitochondrial ADP-ribose Pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801.
- Iordanov, I.; Tóth, B.; Szollosi, A.; Csanády, L. Enzyme activity and selectivity filter stability of ancient TRPM2 channels were simultaneously lost in early vertebrates. eLife 2019, 8, e44556.
- Iordanov, I.; Mihályi, C.; Tóth, B.; Csanády, L. The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity. eLife 2016, 5, e17600.
- Kühn, F.J.P.; Lückhoff, A. Sites of the NUDT9-H Domain Critical for ADP-ribose Activation of the Cation Channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437.
- Perraud, A.-L.; Schmitz, C.; Scharenberg, A.M. TRPM2 Ca2+ permeable cation channels: From gene to biological function. Cell Calcium 2003, 33, 519–531.
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 Functions as a Lysosomal Ca2+-Release Channel in beta cells. Sci. Signal. 2009, 2, ra23.
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sánchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542.
- Du, J.; Xie, J.; Yue, L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc. Natl. Acad. Sci. USA 2009, 106, 7239–7244.
- Kraft, R.; Grimm, C.; Frenzel, H.; Harteneck, C. Inhibition of TRPM2 cation channels by N -(p -amylcinnamoyl)anthranilic acid. J. Cereb. Blood Flow Metab. 2006, 148, 264–273.
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232.
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2019, 85, 102111.
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.-Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, eaav9334.
- Zhang, Z.; Tóth, B.; Szollosi, A.; Chen, J.; Csanády, L. Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. eLife 2018, 7, e36409.
- Csanády, L.; Törőcsik, B. Four Ca2+ Ions Activate TRPM2 Channels by Binding in Deep Crevices near the Pore but Intracellularly of the Gate. J. Gen. Physiol. 2009, 133, 189–203.
- Wang, L.; Fu, T.-M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362, 6421.
- Vilar, B.; Tan, C.-H.; McNaughton, P.A. Heat detection by the TRPM2 ion channel. Nature 2020, 584, E5–E12.
- Kashio, M.; Sokabe, T.; Shintaku, K.; Uematsu, T.; Fukuta, N.; Kobayashi, N.; Mori, Y.; Tominaga, M. Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. USA 2012, 109, 6745–6750.
- Kashio, M.; Tominaga, M. The TRPM2 channel: A thermo-sensitive metabolic sensor. Channels 2017, 11, 426–433.
- Perraud, A.-L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of Free ADP-ribose from Mitochondria Mediates Oxidative Stress-induced Gating of TRPM2 Cation Channels. J. Biol. Chem. 2005, 280, 6138–6148.
- Takahashi, N.; Kozai, D.; Kobayashi, R.; Ebert, M.; Mori, Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011, 50, 279–287.
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the thermal regulation of immunity: The immune system feels the heat. Nat. Rev. Immunol. 2015, 15, 335–349.
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487.
- Fonfria, E.; Murdock, P.R.; Cusdin, F.S.; Benham, C.D.; Kelsell, R.E.; McNulty, S. Tissue Distribution Profiles of the Human TRPM Cation Channel Family. J. Recept. Signal Transduct. 2006, 26, 159–178.
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611.
- Mittal, M.; Nepal, S.; Tsukasaki, Y.; Hecquet, C.M.; Soni, D.; Rehman, J.; Tiruppathi, C.; Malik, A.B. Neutrophil Activation of Endothelial Cell-Expressed TRPM2 Mediates Transendothelial Neutrophil Migration and Vascular Injury. Circ. Res. 2017, 121, 1081–1091.
- Di, A.; Gao, X.-P.; Qian, F.; Kawamura, T.; Han, J.; Hecquet, C.M.; Ye, R.D.; Vogel, S.M.; Malik, A.B. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 2011, 13, 29–34.
- Almasi, S.; Kennedy, B.E.; El-Aghil, M.; Sterea, A.M.; Gujar, S.; Partida-Sánchez, S.; El Hiani, Y. TRPM2 channel–mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J. Biol. Chem. 2018, 293, 3637–3650.
- Zhang, W.; Chu, X.; Tong, Q.; Cheung, J.Y.; Conrad, K.; Masker, K.; Miller, B.A. A Novel TRPM2 Isoform Inhibits Calcium Influx and Susceptibility to Cell Death. J. Biol. Chem. 2003, 278, 16222–16229.
- Miller, B.A.; Hoffman, N.E.; Merali, S.; Zhang, X.-Q.; Wang, J.; Rajan, S.; Shanmughapriya, S.; Gao, E.; Barrero, C.; Mallilankaraman, K.; et al. TRPM2 Channels Protect against Cardiac Ischemia-Reperfusion Injury: Role of Mitochondria. J. Biol. Chem. 2014, 289, 7615–7629.
- Gelderblom, M.; Melzer, N.; Schattling, B.; Göb, E.; Hicking, G.; Arunachalam, P.; Bittner, S.; Ufer, F.; Herrmann, A.M.; Bernreuther, C.; et al. Transient Receptor Potential Melastatin Subfamily Member 2 Cation Channel Regulates Detrimental Immune Cell Invasion in Ischemic Stroke. Stroke A J. Cereb. Circ. 2014, 45, 3395–3402.
- Ye, M.; Yang, W.; Ainscough, J.; Hu, X.-P.; Li, X.; Sedo, A.; Zhang, X.; Chen, Z.; Beech, D.; Sivaprasadarao, A.; et al. TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis. 2014, 5, e1541.
- Zong, P.; Feng, J.; Yue, Z.; Wu, G.; Sun, B.; He, Y.; Miller, B.; Albert, S.Y.; Su, Z.; Mori, Y.; et al. Functional Coupling of TRPM2 and NMDARs exacerbates excitotoxicity in ischemic brain injury. bioRxiv 2021.
- Park, L.; Wang, G.; Moore, J.; Girouard, H.; Zhou, P.; Anrather, J.; Iadecola, C. The key role of transient receptor potential melastatin-2 channels in amyloid-β-induced neurovascular dysfunction. Nat. Commun. 2014, 5, 5318.
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.-F.; LaVine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The Transient Receptor Potential Melastatin 2 (TRPM2) Channel Contributes to β-Amyloid Oligomer-Related Neurotoxicity and Memory Impairment. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 15157–15169.
- Hoffman, N.E.; Miller, B.A.; Wang, J.; Elrod, J.W.; Rajan, S.; Gao, E.; Song, J.; Zhang, X.-Q.; Hirschler-Laszkiewicz, I.; Shanmughapriya, S.; et al. Ca2+ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am. J. Physiol. Circ. Physiol. 2015, 308, H637–H650.
- Düzen, I.V.; Yavuz, F.; Vuruskan, E.; Saracoglu, E.; Poyraz, F.; Göksülük, H.; Candemir, B.; Demiryürek, S. Leukocyte TRP channel gene expressions in patients with non-valvular atrial fibrillation. Sci. Rep. 2017, 7, 1–7.
- Alves-Lopes, R.; Neves, K.B.; Anagnostopoulou, A.; Rios, F.J.; Lacchini, S.; Montezano, A.C.; Touyz, R.M. Crosstalk Between Vascular Redox and Calcium Signaling in Hypertension Involves TRPM2 (Transient Receptor Potential Melastatin 2) Cation Channel. Hypertension 2020, 75, 139–149.
- Zong, P.; Feng, J.; Yue, Z.; Albert, S.Y.; Mori, Y.; Yue, L. TRPM2 deficiency protects against atherosclerosis by inhibiting TRPM2-CD36 inflammatory axis in macrophages. bioRxiv 2021.
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 Channel in Mediating H2O2 -Induced Ca2+ Entry and Endothelial Hyperpermeability. Circ. Res. 2008, 102, 347–355.
- Uchida, K.; Dezaki, K.; Damdindorj, B.; Inada, H.; Shiuchi, T.; Mori, Y.; Yada, T.; Minokoshi, Y.; Tominaga, M. Lack of TRPM2 Impaired Insulin Secretion and Glucose Metabolisms in Mice. Diabetes 2010, 60, 119–126.
- Gao, G.; Wang, W.; Tadagavadi, R.K.; Briley, N.E.; Love, M.I.; Miller, B.A.; Reeves, W.B. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Investig. 2014, 124, 4989–5001.
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17.
- Dardiotis, E.; Aloizou, A.-M.; Markoula, S.; Siokas, V.; Tsarouhas, K.; Tzanakakis, G.; Libra, M.; Kyritsis, A.P.; Brotis, A.; Aschner, M.; et al. Cancer-associated stroke: Pathophysiology, detection and management (Review). Int. J. Oncol. 2019, 54, 779–796.
- Adams, H.P., Jr.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E., 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41.
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458.
- Campbell, B.C.V.; De Silva, D.A.; MacLeod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Prim. 2019, 5, 1–22.
- Marini, C.; De Santis, F.; Sacco, S.; Russo, T.; Olivieri, L.; Totaro, R.; Carolei, A. Contribution of Atrial Fibrillation to Incidence and Outcome of Ischemic Stroke: Results from a Population-Based Study. Stroke 2005, 36, 1115–1119.
- Nattel, S.; Harada, M. Atrial Remodeling and Atrial Fibrillation: Recent advances and translational perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345.
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435.
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.; Helm, R.H. Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes. Circ. Res. 2017, 120, 1501–1517.
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522.
- Hu, Y.-F.; Chen, Y.-J.; Lin, Y.-J.; Chen, S.-A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243.
- Kakae, M.; Miyanohara, J.; Morishima, M.; Nagayasu, K.; Mori, Y.; Shirakawa, H.; Kaneko, S.; Morsishima, M. Pathophysiological role of TRPM2 in age-related cognitive impairment in mice. Neuroscience 2019, 408, 204–213.
- Belrose, J.C.; Xie, Y.-F.; Gierszewski, L.J.; MacDonald, J.F.; Jackson, M.F. Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol. Brain 2012, 5, 11.
- Yue, Z.; Du, J.; Xia, J.; Qin, X.; Zhang, Y.; He, Y.; Fusco, D.; Liang, B.; Yue, L. A Potential Role of TRPM2 Mediated Inflammation in Heart Disease. Circulation 2018, 128 (Suppl. 22).
- Korantzopoulos, P.; Letsas, K.; Fragakis, N.; Tse, G.; Liu, T. Oxidative stress and atrial fibrillation: An update. Free Radic. Res. 2018, 52, 1199–1209.
- Mihm, M.J.; Yu, F.; Carnes, C.; Reiser, P.J.; McCarthy, P.M.; Van Wagoner, D.R.; Bauer, J.A. Impaired Myofibrillar Energetics and Oxidative Injury During Human Atrial Fibrillation. Circulation 2001, 104, 174–180.
- Adam, O.; Lavall, D.; Theobald, K.; Hohl, M.; Grube, M.; Ameling, S.; Sussman, M.A.; Rosenkranz, S.; Kroemer, H.K.; Schäfers, H.-J.; et al. Rac1-Induced Connective Tissue Growth Factor Regulates Connexin 43 and N-Cadherin Expression in Atrial Fibrillation. J. Am. Coll. Cardiol. 2010, 55, 469–480.
- Feng, J.; Armillei, M.K.; Yu, A.S.; Liang, B.T.; Runnels, L.W.; Yue, L. Ca2+ Signaling in Cardiac Fibroblasts and Fibrosis-Associated Heart Diseases. J. Cardiovasc. Dev. Dis. 2019, 6, 34.
- Harada, M.; Luo, X.; Qi, X.Y.; Tadevosyan, A.; Maguy, A.; Ördög, B.; Ledoux, J.; Kato, T.; Naud, P.; Voigt, N.; et al. Transient Receptor Potential Canonical-3 Channel–Dependent Fibroblast Regulation in Atrial Fibrillation. Circulation 2012, 126, 2051–2064.
- Denham, N.; Pearman, C.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.; Trafford, A.W.; Dibb, K.M. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front. Physiol. 2018, 9, 1380.
- Burashnikov, A.; Antzelevitch, C. Reinduction of Atrial Fibrillation Immediately After Termination of the Arrhythmia Is Mediated by Late Phase 3 Early Afterdepolarization–Induced Triggered Activity. Circulation 2003, 107, 2355–2360.
- Qi, X.; Yeh, Y.-H.; Chartier, D.; Xiao, L.; Tsuji, Y.; Brundel, B.J.; Kodama, I.; Nattel, S. The Calcium/Calmodulin/Kinase System and Arrhythmogenic Afterdepolarizations in Bradycardia-Related Acquired Long-QT Syndrome. Circ. Arrhythmia Electrophysiol. 2009, 2, 295–304.
- Hankey, G.J. Potential New Risk Factors for Ischemic Stroke: What is their potential? Stroke 2006, 37, 2181–2188.
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596.
- Howard, V.J.; Madsen, T.E.; Kleindorfer, D.O.; Judd, S.E.; Rhodes, J.D.; Soliman, E.Z.; Kissela, B.M.; Safford, M.M.; Moy, C.S.; McClure, L.A.; et al. Sex and Race Differences in the Association of Incident Ischemic Stroke With Risk Factors. JAMA Neurol. 2019, 76, 179–186.
- Gorgui, J.; Gorshkov, M.; Khan, N.; Daskalopoulou, S.S. Hypertension as a Risk Factor for Ischemic Stroke in Women. Can. J. Cardiol. 2014, 30, 774–782.
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension: The Mosaic Theory and Beyond. Circ. Res. 2021, 128, 847–863.
- Van Varik, B.J.; Rennenberg, R.J.M.W.; Reutelingsperger, C.P.; Kroon, A.A.; De Leeuw, P.W.; Schurgers, L.J. Mechanisms of arterial remodeling: Lessons from genetic diseases. Front. Genet. 2012, 3, 290.
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. In Hypertension Basic Research to Clinical Practice; Springer: Berlin/Heidelberg, Germany, 2016; pp. 511–540.
- Satoh, K.; Nigro, P.; Berk, B.C. Oxidative Stress and Vascular Smooth Muscle Cell Growth: A Mechanistic Linkage by Cyclophilin A. Antioxidants Redox Signal. 2010, 12, 675–682.
- Fortuño, A.; José, G.S.; Moreno, M.U.; Díez, J.; Zalba, G. Oxidative stress and vascular remodelling. Exp. Physiol. 2005, 90, 457–462.
- Sinha, N.; Dabla, P.K. Oxidative Stress and Antioxidants in Hypertension–A Current Review. Curr. Hypertens. Rev. 2015, 11, 132–142.
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020.
- Tran, Q.-K.; Ohashi, K.; Watanabe, H. Calcium signalling in endothelial cells. Cardiovasc. Res. 2000, 48, 13–22.
- Amberg, G.C.; Navedo, M.F. Calcium dynamics in vascular smooth muscle. Microcirculation 2013, 20, 281–289.
- Kuchan, M.J.; Frangos, J.A. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am. J. Physiol. Physiol. 1994, 266, C628–C636.
- Ding, Y.; Vaziri, N.D. Calcium channel blockade enhances nitric oxide synthase expression by cultured endothelial cells. Hypertension 1998, 32, 718–723.
- Batova, S.; Dewever, J.; Balligand, J.-L.; Godfraind, T.; Dessy, C.; Feron, O. The calcium channel blocker amlodipine promotes the unclamping of eNOS from caveolin in endothelial cells. Cardiovasc. Res. 2006, 71, 478–485.
- Resnick, L. The cellular ionic basis of hypertension and allied clinical conditions. Prog. Cardiovasc. Dis. 1999, 42, 1–22.
- Sonkusare, S.; Palade, P.T.; Marsh, J.D.; Telemaque, S.; Pesic, A.; Rusch, N.J. Vascular calcium channels and high blood pressure: Pathophysiology and therapeutic implications. Vasc. Pharmacol. 2006, 44, 131–142.
- Wang, Q.; Guo, W.; Hao, B.; Shi, X.; Lu, Y.; Wong, C.W.; Ma, V.W.; Yip, T.T.; Au, J.S.; Hao, Q.; et al. Mechanistic study of TRPM2-Ca2+-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy 2016, 12, 1340–1354.
- Zhao, Q.; Li, J.; Ko, W.-H.; Kwan, Y.-W.; Jiang, L.; Sun, L.; Yao, X. TRPM2 promotes autophagic degradation in vascular smooth muscle cells. Sci. Rep. 2020, 10, 1–11.
- McCarthy, C.G.; Wenceslau, C.F.; Calmasini, F.B.; Klee, N.S.; Brands, M.W.; Joe, B.; Webb, R.C. Reconstitution of autophagy ameliorates vascular function and arterial stiffening in spontaneously hypertensive rats. Am. J. Physiol. Circ. Physiol. 2019, 317, H1013–H1027.
- Amarenco, P.; Cohen, A.; Tzourio, C.; Bertrand, B.; Hommel, M.; Besson, G.; Chauvel, C.; Touboul, P.-J.T.; Bousser, M.-G. Atherosclerotic Disease of the Aortic Arch and the Risk of Ischemic Stroke. New Engl. J. Med. 1994, 331, 1474–1479.
- Joh, J.H.; Cho, S. Cardiovascular risk of carotid atherosclerosis: Global consensus beyond societal guidelines. Lancet Glob. Health 2020, 8, e625–e626.
- Banerjee, C.; Chimowitz, M.I. Stroke Caused by Atherosclerosis of the Major Intracranial Arteries. Circ. Res. 2017, 120, 502–513.
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721.
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355.
- Heiner, I.; Eisfeld, J.; Warnstedt, M.U.; Radukina, N.; Jüngling, E.; Lückhoff, A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem. J. 2006, 398, 225–232.
- Morad, H.; Luqman, S.; Tan, C.-H.; Swann, V.; McNaughton, P.A. TRPM2 ion channels steer neutrophils towards a source of hydrogen peroxide. Sci. Rep. 2021, 11, 1–16.
- Wittmann, C.; Chockley, P.; Singh, S.K.; Pase, L.; Lieschke, G.; Grabher, C. Hydrogen Peroxide in Inflammation: Messenger, Guide, and Assassin. Adv. Hematol. 2012, 2012, 1–6.
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999.
- Zhang, Z.; Zhang, W.; Jung, D.Y.; Ko, H.J.; Lee, Y.; Friedline, R.H.; Lee, E.; Jun, J.; Ma, Z.; Kim, F.; et al. TRPM2 Ca2+ channel regulates energy balance and glucose metabolism. Am. J. Physiol. Metab. 2012, 302, E807–E816.
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747.
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Clim. Assoc. 2010, 121, 206–220.
- Moore, K.J.; Freeman, M.W. Scavenger Receptors in Atherosclerosis: Beyond lipid uptake. Arter. Thromb. Vasc. Biol. 2006, 26, 1702–1711.
- Baldrighi, M.; Mallat, Z.; Li, X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 2017, 267, 127–138.
- Zhao, M.; Liu, Y.; Wang, X.; New, L.; Han, J.; Brunk, U.T. Activation of the p38 MAP kinase pathway is required for foam cell formation from macrophages exposed to oxidized LDL. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2002, 110, 458–468.
- Ricci, R.; Sumara, G.; Sumara, I.; Rozenberg, I.; Kurrer, M.; Akhmedov, A.; Hersberger, M.; Eriksson, U.; Eberli, F.R.; Becher, B.; et al. Requirement of JNK2 for Scavenger Receptor A-Mediated Foam Cell Formation in Atherogenesis. Science 2004, 306, 1558–1561.
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221.
- Beceiro, S.; Radin, J.N.; Chatuvedi, R.; Piazuelo, M.B.; Horvarth, D.J.; Cortado, H.; Gu, Y.; Dixon, B.; Gu, C.; Lange, I.; et al. TRPM2 ion channels regulate macrophage polarization and gastric inflammation during Helicobacter pylori infection. Mucosal Immunol. 2016, 10, 493–507.
- Knowles, H.; Heizer, J.W.; Li, Y.; Chapman, K.; Ogden, C.A.; Andreasen, K.; Shapland, E.; Kucera, G.; Mogan, J.; Humann, J.; et al. Transient Receptor Potential Melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 2011, 108, 11578–11583.
- Zou, J.; Ainscough, J.F.; Yang, W.; Sedo, A.; Yu, S.-P.; Mei, Z.-Z.; Sivaprasadarao, A.; Beech, D.J.; Jiang, L.-H. A differential role of macrophage TRPM2 channels in Ca2+ signaling and cell death in early responses to H2O2. Am. J. Physiol. Physiol. 2013, 305, C61–C69.
- Duewell, P.; Kono, H.; Rayner, K.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361, Erratum in Nature 2010, 466, 652.
- Chen, R.; Ovbiagele, B.; Feng, W. Diabetes and Stroke: Epidemiology, Pathophysiology, Pharmaceuticals and Outcomes. Am. J. Med. Sci. 2016, 351, 380–386.
- Kissela, B.M.; Khoury, J.; Kleindorfer, D.; Woo, D.; Schneider, A.; Alwell, K.; Miller, R.; Ewing, I.; Moomaw, C.J.; Szaflarski, J.P.; et al. Epidemiology of Ischemic Stroke in Patients With Diabetes: The greater Cincinnati/Northern Kentucky Stroke Study. Diabetes Care 2005, 28, 355–359.
- Gæde, P.; Lund-Andersen, H.; Parving, H.-H.; Pedersen, O. Effect of a Multifactorial Intervention on Mortality in Type 2 Diabetes. New Engl. J. Med. 2008, 358, 580–591.
- A DiMeglio, L.; Evans-Molina, C.; Oram, R. Type 1 diabetes. Lancet 2018, 391, 2449–2462.
- Grodsky, G.M.; Bennett, L.L. Cation Requirements for Insulin Secretion in the Isolated Perfused Pancreas. Diabetes 1966, 15, 910.
- Gromada, J.; Høy, M.; Renström, E.; Bokvist, K.; Eliasson, L.; Göpel, S.; Rorsman, P. CaM kinase II-dependent mobilization of secretory granules underlies acetylcholine-induced stimulation of exocytosis in mouse pancreatic B-cells. J. Physiol. 1999, 518, 745–759.
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815.
- Bari, M.R.; Akbar, S.; Eweida, M.; Kühn, F.J.; Gustafsson, A.J.; Lückhoff, A.; Islam, S. H2O2-induced Ca2+ influx and its inhibition by N-(p-amylcinnamoyl) anthranilic acid in the β-cells: Involvement of TRPM2 channels. J. Cell. Mol. Med. 2009, 13, 3260–3267.
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82.
- Espinoza-Jiménez, A.; Navarrete-Peón, A.; Terrazas, L.I. Alternatively Activated Macrophages in Types 1 and 2 Diabetes. Mediat. Inflamm. 2012, 2012, 815953.
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative stress and beta-cell dysfunction. Pflug. Arch. 2010, 460, 703–718.
- Domínguez, C.; Ruiz, E.; Gussinye, M.; Carrascosa, A. Oxidative Stress at Onset and in Early Stages of Type 1 Diabetes in Children and Adolescents. Diabetes Care 1998, 21, 1736–1742.
More