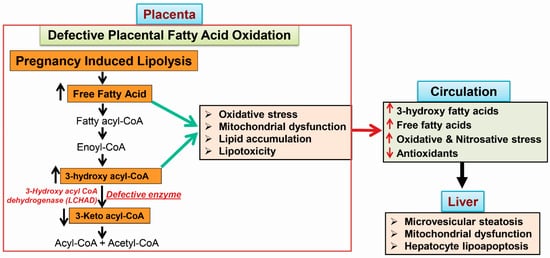
Acute fatty liver of pregnancy (AFLP), a catastrophic illness for both the mother and the unborn offspring, develops in the last trimester of pregnancy with significant maternal and perinatal mortality. AFLP is also recognized as an obstetric and medical emergency. Maternal AFLP is highly associated with a fetal homozygous mutation (1528G>C) in the gene that encodes for mitochondrial long-chain hydroxy acyl-CoA dehydrogenase (LCHAD). The mutation in LCHAD results in the accumulation of 3-hydroxy fatty acids, such as 3-hydroxy myristic acid, 3-hydroxy palmitic acid and 3-hydroxy dicarboxylic acid in the placenta, which are then shunted to the maternal circulation leading to the development of acute liver injury observed in patients with AFLP.
- acute fatty liver of pregnancy
- 3-hydroxy fatty acids
- lipoapoptosis
- fatty acid oxidation
1. Maternal Liver Disease Associated with Fatty Acid Oxidation Defects
1.1. Acute Fatty Liver of Pregnancy
1.2. The Incidence of AFLP and LCHAD Mutations
1.3. 3-Hydroxy Fatty Acid Accumulation
2. Metabolic Phenotypes Associated with 3-Hydroxy Fatty Acid Accumulation
2.1. Hypoglycemia in AFLP and Hormonal Regulation
2.2. Mitochondrial Trifunctional Protein (MTP)-Deficient Mice Develop Intra Uterine Growth Retardation (IUGR), Neonatal Hypoglycemia, and Sudden Death
2.3. MTP Heterozygous Mice Develop Hepatic Insulin Resistance
3. Mechanisms of 3-Hydroxy Fatty Acid-Induced Lipotoxicity
3.1. Placental Damage in AFLP Patients
3.2. Subcellular Damage and Oxidative Injury
3.3. 3-Hydroxy Fatty Acid-Induced Hepatocyte Lipoapoptosis
Abbreviations
| AFLP | acute fatty liver of pregnancy |
| ALT | alanine amino transferase |
| AST | aspartate amino transferase |
| ALP | alkaline phosphatase |
| CPT2 | carnitine palmitoyl transferase 2 |
| DHA | docosa hexaenoic acid |
| FoxO | forkhead family of transcription factor class O |
| GGT | γ-glutamyl transpeptidase |
| LCHAD | long chain hydroxy acyl-CoA dehydrogenease |
| MAPK | mitogen activated protein kinases |
| MTP | mitochondiral trifunctional protien |
| PUMA | p35-upregulated modulator of apoptosis |
| TCA cycle | tricarboxylic acid cycle |
| 3-HFA | 3-hydroxy fatty acid |
| 3-HMA | 3-hydroxy myristic acid |
| 3-HOA | 3-hydroxy octanoic acid |
| 3-HPA | 3-hydroxy palmitic acid |