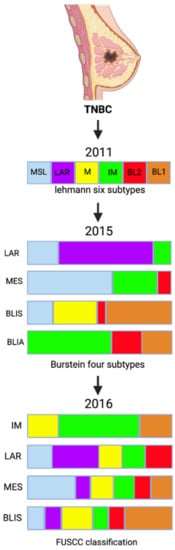
In view of the emerging important role of long noncoding RNAs (lncRNAs) in cellular processes, a new classification incorporating both messenger RNA (mRNA) and lncRNA transcriptome profiles was suggested to help provide a better understanding of the heterogeneity of TNBC (
Figure 1). In 2016, Liu et al. performed a categorization analysis of 165 TNBC samples that combined mRNA expression analysis and co-expression network analysis, aiming to identify interactions between mRNAs and lncRNAs
[11]. They also investigated IM subtype genes, previously linked to the stimulation of T-cells and to innate and regular immune responses (
Table 1)
[6]. A strong association was identified between immune cell processes and TNBC tumorigenesis. IM subtype genes were identified to engage in regulating immune cells through their modulation of cytokine signaling, antigen processing, and immune cell signaling pathways involving T-cells, B-cells, chemokines and the nuclear factor-κB (NF-κB)
[11]. Receptor-interacting protein 2 (RIP2) has been linked to chemoresistance of TNBC against paclitaxel. Jaafar et al. demonstrated that high expression of RIP2 correlated with a worse prognosis and a higher risk of recurrence since RIP2 lead to NF-κB activation, which contributed to higher expression of pro-survival proteins and cell survival
[12]. Other genes, known to impact immune response—such as C-C motif chemokine teceptor-2 (CCR2), chemokine ligand 5 (CCL5), cluster of differentiation 1 (CD1C), C-X-C motif chemokine ligand 10 (CXCL10), C-X-C motif chemokine ligand 11 (CXCL11), and C-X-C motif chemokine ligand 13 (CXCL13)—were also expressed in the TNBC IM subtype, further confirming the role of immunity in TNBC IM tumorigenesis.
Figure 1. TNBC classification over the years.
Table 1. TNBC subtypes based on the FUSCC (Fudan University Shanghai Cancer Center) classification criteria
[6].
| FUSCC Classification |
Pathways |
| IM (immunomodulatory) |
↑ |
|
-
Cytokine–cytokine receptor interaction
-
T cell receptor signaling pathway
-
B cell receptor signaling pathway
-
Chemokine signaling pathway
-
NF-kappa-B signaling pathway
|
|
| LAR (luminal androgen receptor) |
↑ |
| |
,22,23]. Two large, randomized trials—CALGB 40603/Alliance trial and GeparSixto—compared conventional chemotherapy regimens with or without added Carboplatin and showed higher pCR rates with inclusion of the platinum-based agent. The CALGB 40603/Alliance trial assessed the value of adding Bevacizumab +/− Carboplatin to neoadjuvant chemotherapy in stage II and III TNBC in 443 patients
[24]. The proportion of patients who attained pCR increased remarkably from 41% to 54% with the use of Carboplatin (OR = 1.71;
p = 0.0029). The long-term OS was not powered in the trial, and the addition of Carboplatin to conventional chemotherapy did not increase long-term OS
[25]. The GeparSixto trial involved 595 patients diagnosed with stages II or III TNBC, who were randomized to receive either Carboplatin or no Carboplatin with a backbone regimen of Paclitaxel, liposomal Doxorubicin, and Bevacizumab
[26]. The pCR rates were considerably higher in the carboplatin group: 53.2% vs. 36.9 (
p = 0.005) (
Table 2). The result of a meta-analysis looking at 9 randomized controlled trials (RCTs) (
n = 2109) revealed that adding platinum to neoadjuvant chemotherapy considerably improved pCR rate from 37.0% to 52.1% (OR 1.96, 95% confidential interval (CI) 1.46–2.62,
p < 0.001)
[27]. Loibl et al. presented their updates from the BrighTNess trial, a randomized phase III clinical trial, with three treatment arms and a total of 634 patients with TNBC: the established neoadjuvant regimen consisting of Paclitaxel alone (P) (
n = 158), Paclitaxel and Carboplatin alone (PCb) (
n = 160), and Paclitaxel, Carboplatin and the PPAR inhibitor Veliparib (PCbV) (
n = 316). Event-free survival, OS, and safety outcomes were assessed with a ≥4 years of follow-up period
[28]. pCR was significantly improved when Carboplatin was added, with or without the addition of Veliparib, to Paclitaxel-based neoadjuvant chemotherapy. Also, adding Carboplatin to Paclitaxel improved pCR and EFS without increasing myelodysplastic syndrome or acute myeloid leukemia
[28]. When compared to P alone, HR for EFS with PCbV was 0.63 (95% CI: 0.43–0.92,
p = 0.016), and HR for EFS with PCb was 0.57 (95% CI 0.36–0.91,
p = 0.018)
[28]. Based on the latest American Society of Oncology (ASCO) recommendations, carboplatin may be offered to patients with TNBC to increase pathologic complete response
[29].
Table 2. Frequencies of pCR from clinical trials involving carboplatin.
Trials
(References) |
Regimen 1 (R1) |
Nb. of Patients |
Regimen 2 (R2) |
Nb of Patients |
pCR Rate (R1 vs. R2) |
p-Value |
| GeparOcto GBG84 [30] |
P | + | NPLD | + | Cb | ;
q1w for 18 weeks |
203 |
E | then | P | then | C | ;
q2w/3 cycles over 18 weeks |
200 |
51.7% vs. 48.5% |
0.518 |
| GALGB40603 Alliance [24] |
( | P | q1w for 12 weeks then | ddAC | q2w/4 cycles) + ( | Cb | q3w/4 cycles ± | Bev | |
|
| . q2w/9cycles) |
221 |
P | q1w for 12 weeks then | ddAC | q2w/4 cycle |
MES (mesenchymal-like) |
↑ |
| |
|
| BLIS (basal-like and immune suppressed) |
↑ |
| |
|
| ↓ |
| |
|
Conversely, tumorigenesis in the LAR subtype is closely related to hormonal regulation and activity, particularly the metabolism of androgen, chlorophyll, estrogen, and porphyrin, as well as the biosynthesis of hormones. LAR cells also show an increased expression of the peroxisome proliferator-activated receptor-γ (PPAR-γ). PPAR-γ is implicated in tumor cell proliferation, growth invasion, and phenotypic changes in differentiation status, but also correlates with quantitative androgen receptor expression, a defining feature of LAR
[13]. Interestingly, despite lack of ER expression on its cell surface, LAR is clinically responsive to both anti-estrogen and anti-androgen therapy. This can be explained by the positive molecular activity of the estrogen receptor signaling pathway implicated in LAR tumorigenesis, despite LAR cells being ER-negative
[10].
On the other hand, the MES subtype harbors a unique gene ontology, characterized by the interaction between extracellular matrix receptors, gap junction transmembrane channels, the transforming Growth Factor-β (TGF-β) signaling pathway, and growth factor-associated pathways, notably the adipokine pathway and ATP-binding cassette (ABC) transporters pathway
[14].
Furthermore, the BLIS subtype is distinguished by a pathogenesis that strongly implicates cell cycle and resultant cell division processes in addition to DNA repair, replication, and regulation mechanisms. BLIS cells show increased quantitative expression of genes involved in cell proliferation such as the mitotic checkpoint serine/threonine-protein kinase budding uninhibited by benzimidazoles 1 (BUB1), and the protein coding genes centromere protein F (CENPF) and protein regulator of cytokinesis 1 (PRC1). This translates into a highly proliferative clinical nature of BLIS tumors
[11], further allowed by the downregulation of immunologic processes specifically involving T-cell signaling, B-cell activation and dendritic cells chemotaxis. These molecular processes translate into shorter relapse-free survival (RFS) and increased recurrence rate on the clinical level, supporting previous findings by Burstein et al.
[11].
Although progress in next generation sequencing has facilitated unraveling potentially actionable targets, not many findings have not been translated into daily clinical practice due to limited benefit from targeted therapy observed in clinical trials for unselected TNBC patients. The molecular subtyping enables the identification of molecularly homogenous groups with enrichment of certain genomic alterations. This paves the way for effective methods for drug development using subtype-specific clinical investigations. A precision medicine paradigm in the context of transcriptomic subtyping should be developed and fine-tuned for patients with TNBC.
3. Chemotherapy for Triple Negative Breast Cancer
TNBC has historically had limited treatment options when compared to other types of BC. The mainstay of treatment for TNBC remains cytotoxic chemotherapy, despite the emergence of new biologic and targeted agents. The therapeutic benefits of cytotoxic chemotherapy in TNBC are well established, with comprehensive data on the efficacy of chemotherapy in the neoadjuvant, adjuvant, and metastatic settings. Compared with hormone receptor-positive (HR+) BC, the use of chemotherapy regimens in the neoadjuvant treatment of TNBC has a significantly higher pathological response rate and can considerably ameliorate the prognosis of TNBC patients
[15]. Nevertheless, TNBC carries an overall inferior prognosis despite its chemo-sensitivity
[16]. The use of neoadjuvant systemic treatment (NST) in the early stages is becoming the standard of care in TNBCs and is associated with higher pathological complete response (pCR) rates (30–40%) as compared to other BC subtypes
[17]. Patients who achieve pCR with primary therapy have improved survival outcomes
[18]. As such, pCR is predictive of improved long-term outcomes for TNBC and is a reliable endpoint in clinical trials evaluating the efficacy of neoadjuvant chemotherapy.
Adriamycin, cyclophosphamide, and paclitaxel combinations are considered to be standard neoadjuvant chemotherapy regimen against TNBC and result in pCR rates of 35–45%
[19]. The addition of platinum-based chemotherapy has been proposed. Despite improved short term pCR rates, long term outcomes remain unknown
[20]. The systemic chemotherapy regimens options for TNBC recommended by National Comprehensive Cancer Network (NCCN) guidelines include the following: Docetaxel and Cyclophosphamide (TC), Taxel/Docetaxel, Adriamycin, and Cyclophosphamide (TAC), Adriamycin and Cyclophosphamide (AC), Cyclophosphamide, Methotrexate, and Fluorouracil (CMF), Cyclophosphamide, Adriamycin, and Fluorouracil (CAF), and Cyclophosphamide, Epirubicin, Fluorouracil and Paclitaxel/Docetaxel (CEF-T). These DNA damaging agents show increased activity in cancers with a germline BRCA mutation, as BRCA 1/2 proteins play an essential role in repairing DNA damage
[21].
TNBC is also highly sensitive to platinum salts because a high proportion of these tumors exhibit BRCA-like status
[20][22][23][20| 212 |
| 54% vs. 41% |
0.0029 |
| GeparSixto GBG66 [26] |
(P q1w for 18 weeks + NPLD q1w for 18 weeks + Bev. q3w/6 cycles) + Cb q1w for 18 weeks |
158 |
P | q1w for 18 weeks + | NPLD | q1w for 18 weeks + | Bev | . q3w/6 cycles |
157 |
53.2% vs. 36.9% |
0.005 |
| Zhang et al. [31] |
P | + | Cb | q3w/4–6 cycles |
47 |
P | + | E | q3w/4–6 cycles |
44 |
38.6% vs. 14.0% |
0.014 |
| Ando et al. [32] |
( | P | q2w/2 cycles then | CEF | q2w/4 cycles) + | Cb | q3w/4 cycles |
37 |
P | q2w/2 cycles then | CEF | q2w/4 cycles |
38 |
61.2% vs. 6.3% |
0.003 |
P = Paclitaxel; NPLD = Nonpegylated Liposomal Doxorubicin; Cb = Carboplatin; E = Epirubicin; C = Cyclophosphamide; ddAC = Doxorubicin plus Cyclophosphamide; Bev. = Bevacizumab; CEF = Cyclophosphamide plus Epirubicin plus 5-fluorouracil.
4. Detecting PDL-1 Expression in TNBC
As the importance of immunotherapies targeting PD-1/ PD-L1 is evolving, concerns are arising regarding diagnostic tests that detect the level of these molecules and thus predict outcomes in cancer patients. Routinely, immunohistochemistry is used to measure PD-L1 expression. Currently, many of the commercially available tests are designed by antibody clones that detect the presence of the PD-L1 protein. Moreover, multiple expression scores and cutoffs exist
[33]. Of the relevant PD-L1 scores are the tumor cell score, tumor proportion score, the immune cell score, and the combined positive score
[34][35][34,35].
There are four PD-L1 immunohistochemical (IHC) assays registered with the FDA, using four different PD-L1 antibodies (22C3, 28–8, SP263, SP142), on two different IHC platforms (Dako and Ventana), each with their own scoring systems
[36]. Attempts at harmonization of PD-L1 IHC antibodies and staining platforms are underway
[37]. While PD-L1 IHC can be used to predict likelihood of response to anti-PD-1 or anti-PD-L1 therapy, a proportion of patients that are negative can have response and identification of alternative biomarkers is critical to further refine selection of patients most likely to respond to these therapies
[36][38][39][40][36,38,39,40].