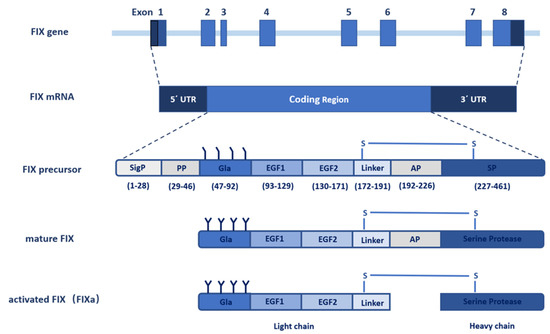
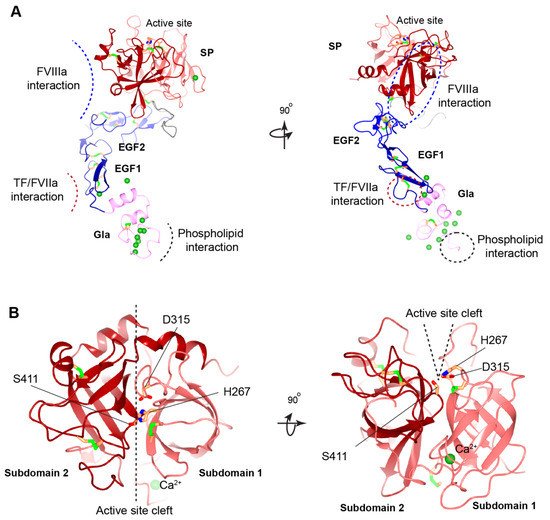
Figure 3. Structural model of human FIXa. (
A) Overview of human FIXa’s structural model. The UniProt accession number of human FIX protein is P00740. The model is constructed in the following ways. The overall architecture is generated by Alphafold prediction
[46][36]. Next, three partial human crystal structures, Gla domain (PDB: 1NL0)
[42][25], EGF1 domain (PDB: 1EDM)
[47][37], and combining EGF2-SP domain (PDB: 2WPH)
[48][38] are superimposed onto the Alphafold model and a porcine FIXa crystal structure (PDB: 1PFX)
[49][39] is superimposed to correct the inter-domain orientation between heavy and light chains. These superimposed crystal structures are combined to generate the final model. The Gla domain, EGF domains, SP domain, and linker sequence are colored in pink, blue, red, and grey, respectively. The Ca
2+ ions are shown in the green sphere, and disulfide bonds in the green bars. The predicted interaction interfaces in FIXa with FVIIIa, with TF/FVIIa, and with membrane phospholipid are indicated by dashed curves (
left) or dashed circles (
right). The ω-loop in the Gla domain is also indicated by a dashed circle (
right). (
B) Structural model of human FIXa’s SP domain. Subdomain 1 and subdomain 2 are colored in light red and dark red, respectively. The active site cleft is formed between the two subdomains and side chains of the catalytic triad (His267, Asp315, and Ser415) are shown. These figures are generated with CCP 4 mg
[50][40].
3.2.5. Missense Mutations in EGF1 and EGF2 Domains
Disulfide bonds maintain the structural integrity of the EGF domains (
Figure 3A). Missense mutations of the cysteines break the disulfide bonds and cause a severe bleeding phenotype
[16][41]. These cysteine mutations in hemophilia B patients are associated with reduced FIX antigen level, suggesting that they destabilize the FIX
[16][41]. Besides the disulfide bonds, Ca
2+ ion binding to the EGF1 domain is essential for stabilizing its conformation (
Figure 3A) and for the assembly of the Xase complex on the phospholipid surface. Asp93, Gln96, and Asp110 in the EGF1 domain are designated for Ca
2+ ion binding
[47][37]. Missense hemophilic mutations in these residues
[16][51][52][41,83,84] may disturb the stability of EGF1 domain, which serves to correctly position the SP domain for optimal interaction with FVIIIa
[53][44].
Generating the full enzymatic activity for FIXa requires its binding to FVIIIa. Several hemophilic mutations in EGF domains may interfere with FIXa binding to FVIIIa
[41][21][34,66]. A model of the Xase complex suggested that the conserved residues, Ile112, Tyr115, and Trp118, in the EGF1 domain, and Ile136, Asn138, and Arg140 in the EGF2 domain contribute to FVIIIa binding
[49][39]. However, mutations in the EGF2 domain mainly affect FIXa binding to FVIIIa by hindering the interaction of FIXa’s SP domain with FVIIIa
[53][44]. Additionally, hemophilia B patients with missense mutations in residues Ile136 and Val153 of the EGF2 domain may disrupt the interaction of FIXa with the activated platelet surface and the assembly of the Xase complex on activated platelets.
In the extrinsic coagulation pathway, activation of FIX is triggered by its interaction with the TF/FVIIa complex. A study showed that FIX are barely activated in hemophilia B patients with p.Gly94Arg and p.Gly94Val mutations that disrupt the interaction of FIX with TF/FVIIa complex
[54][85], indicating that the EGF1 domain is essential for this interaction. In a binding model of FIX/TF/FVIIa, Asp95, Glu98, Ser99, Asn104, Phe123, Asn127, and Glu129 on the surface of the EGF1 domain were proposed to be the key determinants for FIX binding to TF
[40][32] (
Figure 3A). Consistent with this model, numerous missense mutations in these residues have been reported to cause hemophilia B
[16][41]. Experimental evidence, however, is required to establish the role of these residues in TF binding.
3.2.6. Missense Mutations at Cleavage Site of Activation Peptide
More than 300 hemophilia B patients in the
F9 database have been reported with mutations at the cleavage site of activation peptide, namely at residues Arg191 or Arg226
[16][55][41,86]. However, hemophilia B patients are rarely reported to have mutations in the other residues of the activation peptide, and only seldom have polymorphisms been retrieved. Removal of the activation peptide requires cleavages at both the Arg191 and Arg226 site, and the missense mutations disrupt these cleavages
[55][86]. Seminal studies showed that the cleavage only at Arg191 resulted in the loss of FIXa catalytic activity
[56][57][26,87], whereas the cleavage only at Arg226 retains the catalytic activity but results in suboptimal binding to FVIIIa
[58][55][48,86]. Consistently, most patients with missense mutations at Arg191 are associated with a moderate or mild bleeding tendency
[16][59][60][61][41,88,89,90], whereas those at Arg226 have a severe bleeding phenotype
[16][62][63][64][41,91,92,93]. In contrast, the p.Arg226Lys mutation that retains a positively charged residue does not appear to cause hemophilia B, as the variant does not hinder the release of the activation peptide
[55][86].
Patients with mutations at Arg191 and Arg226 have different FIX antigen levels. Mutations at Arg226 residues are associated with normal or increased FIX levels, whereas Arg191 mutations are characterized by normally or modestly reduced antigen levels
[55][86]. These observations show the detrimental effects of Arg191 mutations on the protein folding, secretion, or scavenging processes
[6], all of which contribute to decreased antigen levels in the FIX. In turn, these features shape the bleeding phenotypes of patients, and produce the graded moderate to mild bleeding severity in hemophilia B patients with the Arg191Cys>Leu>Pro>His mutations
[55][86].
3.2.7. Missense Mutations in SP Domain
Among the hemophilia B patients with missense mutations, more than 55% have missense mutations located in the SP domain of FIX
[9][16][9,41], emphasizing the importance of this domain. Cysteine residues involved in disulfide bond formation in the SP domain are important for stabilizing the protein folding (
Figure 3), and missense mutations in these residues cause severe hemophilia B. The SP subdomain 1 contains a high affinity Ca
2+ ion binding site (277–311 aa) at a surface loop
[49][39] (
Figure 3). Mutations in Arg294, Arg298, and Asn310 of the Ca
2+ ion binding loop show significantly decreased FIX antigen levels
[16][65][66][67][41,94,95,96], implying that the calcium loop affects the stability of the SP domain.
Impaired interaction with FVIIIa is another mechanism causing hemophilia B for missense mutations in the SP domain, especially those in subdomain 2
[16][41]. The 378-helix in SP subdomain 2 was thought to bind to FVIIIa
[41][34] (
Figure 3A). All known sequences from different species in this surface-exposed helix of FIXa are identical, and missense mutations in eight of the nine residues in this region cause hemophilia B
[68][97], because mutations in this helix reduce FIXa’s affinity to FVIIIa. In addition, residues Lys339 in 339-helix and Asn392 were proposed to bind the A2 subunit of FVIIIa
[49][39]. Subdomain-2 also contains an autolysis loop (356–369 aa), and mutations in this loop result in diminished binding of FIXa to FVIIIa. For example, mutation at Lys362 results in a substantial reduction in the rate of FX activation in the presence and absence of FVIIIa
[69][98].
Additionally, the cleft between both SP subdomains forms the active site pocket that contain the catalytic triad of His267, Asp315, and Ser411
[49][39] (
Figure 3B). Hemophilia B associated missense mutations in and around the active site pocket impairs the active site formation or substrate recognition
[16][41].