Lipid transfer proteins (LTPs) are recognized as key players in the inter-organelle trafficking of lipids and are rapidly gaining attention as a novel molecular target for medicinal products. In mammalian cells, ceramide is newly synthesized in the endoplasmic reticulum (ER) and converted to sphingomyelin in the trans-Golgi regions. The ceramide transport protein CERT, a typical LTP, mediates the ER-to-Golgi transport of ceramide at an ER-distal Golgi membrane contact zone. About 20 years ago, a potent inhibitor of CERT, named (1R,3S)-HPA-12, was found by coincidence among ceramide analogs. Since then, various ceramide-resembling compounds have been found to act as CERT inhibitors. Nevertheless, the inevitable issue remains that natural ligand-mimetic compounds might directly bind both to the desired target and to various undesired targets that share the same natural ligand. To resolve this issue, a ceramide-unrelated compound named E16A, or (1S,2R)-HPCB-5, that potently inhibits the function of CERT has recently been developed, employing a series of in silico docking simulations, efficient chemical synthesis, quantitative affinity analysis, protein–ligand co-crystallography, and various in vivo assays. (1R,3S)-HPA-12 and E16A together provide a robust tool to discriminate on-target effects on CERT from off-target effects.
1. Introduction
Lipids are the major constituents of all cell membranes and play dynamic roles in organelle structure and function. In eukaryotic cells, the endoplasmic reticulum (ER) is the main center for the synthesis of diverse types of lipids. Lipids newly synthesized in the ER are rapidly and accurately delivered to other organelles by a variety of lipid transfer proteins (LTPs), in a nonvesicular manner, at zones where the ER is in contact with other specific organelles
[1][2][3][4][5][6][1,2,3,4,5,6]. Various viruses and obligate intracellular bacteria hijack L
ipid transfer proteins (LTPs
) of host cells for their proliferation
[7][8][9][10][7,8,9,10]. Therefore, LTPs are fundamental for the metabolism of lipids and biogenesis of membrane compartments in cells and have been gaining attention as a novel type of molecular medicine target. Nonetheless, the repertoire of specific LTP inhibitors to date is limited.
Sphingolipids are a class of lipids that contain the long-chain amino alcohols termed sphingoid bases, including sphingosine, dihydrosphingosine, and phytosphingosine, as their structural backbone (
Figure 1A). Sphingolipids are ubiquitous constituents of cell membranes in eukaryotes and also present in a limited number of prokaryote species
[11][12][13][14][11,12,13,14]. Sphingolipids participate in various biological events, including cell growth, apoptosis, differentiation, and adhesion (reviewed in
[15][16][15,16]). The choline-containing sphingophospholipid sphingomyelin is a ubiquitous and predominant type of sphingolipid found in mammals and is thought to be a chemically robust but noncovalently interactive type of phospholipid, unlike other types of phospholipid
[11].
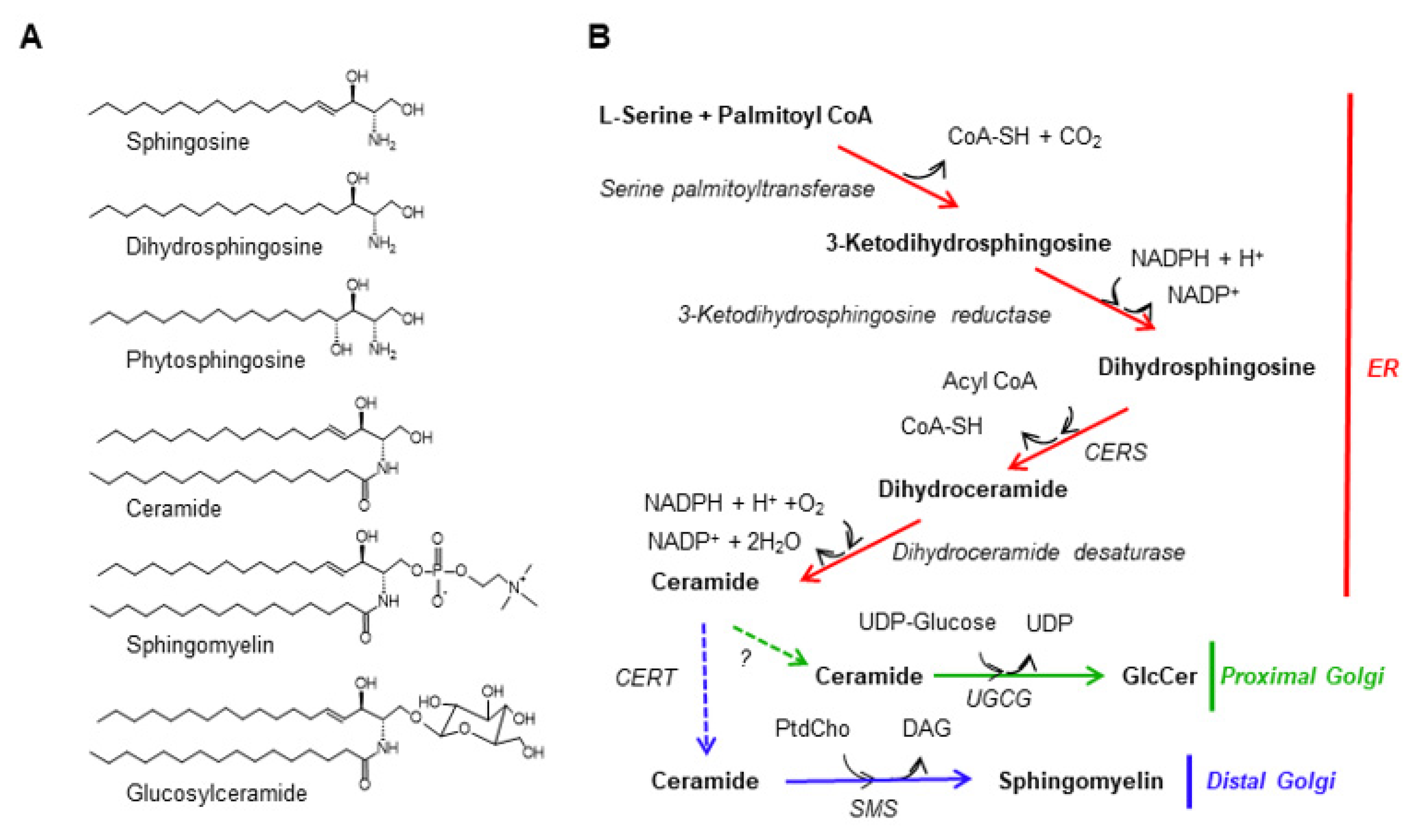 Figure 1.
Figure 1. The de novo synthetic pathway for sphingomyelin. (
A) Structure of mammalian sphingolipids. The chemical structures of three major sphingoid bases and several complex sphingolipids in mammalian cells are shown. Ceramide, sphingomyelin, and GlcCer are depicted as the
N-palmitoyl sphingosine type, although there are various combinations of sphingoid bases and
N-acyl chains in cells. (
B) The de novo synthetic pathways for sphingomyelin and GlcCer in mammalian cells are shown. Solid arrows represent enzymatic reactions while dotted arrows represent inter-organelle transport processes. PtdCho, phosphatidylcholine.
In mammalian cells, sphingomyelin is mainly located in the plasma membrane (PM), although the Golgi apparatus (where sphingomyelin is synthesized) and endosomes/lysosomes (to which the PM-derived endocytosis is directed) also have sphingomyelin
[17][18][17,18]. In the PM phospholipid bilayer, sphingomyelin is predominantly distributed in the exoplasmic leaflet, with less found in the cytoplasmic leaflet. Cytoplasm-oriented sphingomyelin was recently shown to be pivotal for forming a PM-derived, concave, tubular structure
[19]. Sphingomyelin, in concert with cholesterol, generates nano-scale membrane domains (so-called “lipid rafts” or “detergent-resistant membranes”), in which the spatiotemporal confinement of various molecules needed for signaling and other dynamic events of membranes can efficiently occur (see
[20][21][20,21] for recent reviews). Sphingomyelin may interact with specific membrane proteins as their functional modulator, while sphingomyelin is also an important metabolic reservoir for sphingolipid mediators such as ceramide, sphingosine, and sphingosine-1-phosphate, which are produced as catabolites of sphingomyelin (reviewed in
[15][22][23][24][15,22,23,24]).
The ceramide transport protein CERT, a typical LTP, mediates the transport of ceramide from the ER to the Golgi apparatus, where ceramide is converted to sphingomyelin
[11][25][11,25]. CERT was first discovered in 2003. In 2001, a ceramide-mimetic compound that potently inhibits ER-to-Golgi transport of ceramide in cells was serendipitously developed
[26]; it was later shown to be a selective inhibitor of CERT
[27]. Nevertheless, there was inevitable concern that natural ligand-mimetic compounds may directly bind to the desired target but also to various undesired targets that share the same natural ligand. Hence, ceramide-nonmimetic inhibitors have recently been developed using a structure-based drug design approach
[28]. The establishment of a set of ligand-mimetic and nonmimetic inhibitors has provided a pharmacological tool to discriminate on-target effects from off-target effects.
2. Possible Applications of CERT Inhibitors
CERT plays a key role in the ceramide-sphingomyelin metabolic axis (reviewed in
[11]). As more evidence has accumulated that ceramide and sphingomyelin participate in various physiological and pathophysiological events
[15][16][23][24][15,16,23,24], more attention has been paid to CERT. Accordingly, it has been clarified that CERT and CERT
L (also known as GPBPΔ26 and GPBP, respectively) participate in various events and functions in mammalian cells, including polyploid cancer cell death
[29][95], EGF receptor signaling
[30][96], lipotoxicity and glucolipotoxicity in islet β-cells
[31][32][97,98], muscle insulin signaling
[33][99], myofibril formation
[34][100], integrity of glomerular basement membrane
[35][101], cytotoxic autophagy
[36][102], mitochondrial maintenance
[37][103], and senescence
[38][104].
Interest in CERT inhibitors has also been increasing, although
itas far seems thatas we know CERT inhibitors have yet to be used clinically. One of the most desirable applications of CERT inhibitors may be their therapeutic use in cancer, because the upregulation of intracellular ceramide levels by the blockage of the ceramide-to-sphingomyelin conversion through the use of CERT inhibitors would facilitate the efficacy of existing anti-cancer drugs, as previously proposed
[29][95]. For further possible applications of CERT inhibitors in the treatment of cancer, see the recent review by Chung et al.
[39][88].
Another important application may be the therapeutic or prophylactic use of CERT inhibitors to treat disorders caused by abnormally activated CERT. Rapid advances in human clinical genetics studies have revealed that various specific missense mutations in the human
CERT1 gene are associated with hereditary intellectual disabilities and mental development disorders with an autosomal dominant inheritance pattern
[40][41][42][43][44][44,45,46,47,48], and some of these mutations were recently shown to compromise the functional repression of CERT
[44][45][48,49]. Thus, a CERT inhibitor could be a rational tool to reduce abnormally activated CERT to a normal level. This could in turn improve the pathology of the
CERT1 mutation-caused disease without shutting off the physiologically indispensable function of CERT, although more studies will be necessary to identify a clinically usable CERT inhibitor.
CERT1 encodes at least two CERT-related proteins, CERT/GPBPΔ26 and CERT
L/GPBP, which are transcribed from different splicing transcripts. Revert et al. showed that CERT
L/GPBP, but not CERT/GPBPΔ26, is exported from cells via a nonconventional secretion pathway
[46][52]. Intriguingly, CERT
L/GPBP was reported to be capable of binding various extracellular proteins including type IV collagen
[47][50], serum amyloid P-component
[48][105], the complement factor C1q
[49][106], and the amyloid precursor protein and amyloid-β
[50][65]. Thus, CERT inhibitors may also be a useful tool to investigate the physiological and pathophysiological significance of interactions between CERT
L/GPBP and extracellular proteins, although it remains unclear whether the function of extracellular CERT
L/GPBP is relevant to its ceramide-binding activity. Crivelli et al. recently revealed that genetically modified enhancement of CERT
L expression in the brain in a familial Alzheimer’s disease model in mice reduces the amyloid-β level, accompanied by a decreasing ceramide level concomitant with increasing sphingomyelin level in the brain; they also found that administration of HPA-12 to the model mice for 4 weeks exacerbated the ceramide level and amyloid-β pathology
[50][65]. Thus, long-term administration of a CERT inhibitor might aggravate Alzheimer’s disease. If ceramide-binding activity is also crucial to the function of extracellular CERT
L/GPBP, then membrane-impermeable compounds, rather than membrane-permeable compounds, will be more appropriate to selectively modulate their extracellular function(s) without inhibiting their intracellular function(s).
The pharmacological or genetic inhibition of CERT negatively affects the proliferation of several types of intracellular pathogens, including hepatitis C virus
[51][52][53][107,108,109], rubella virus
[54][110], and the obligate intracellular bacterium Chlamydia
[9][55][56][9,111,112]. Hence, CERT inhibitors might be applicable as anti-infectious disease agents, although pathogen genome-encoded factors, rather than human genome-encoded factors, may be more suitable molecular targets of anti-infectious disease drugs in terms of the risk of side effects.