Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Amina Yu and Version 1 by Antonino Tuttolomondo.
Diabetes mellitus is a comprehensive expression to identify a condition of chronic hyperglycemia whose causes derive from different metabolic disorders characterized by altered insulin secretion or faulty insulin effect on its targets or often both mechanisms . Atherosclerosis (ATS) is the most frequent cause of arterial vasculopathy and is undoubtedly an insidious condition: it is unlikely to be the trigger in coronary artery disease, ischemic stroke, and peripheral artery disease (PAD) on its own; instead, it acts together with other chronic degenerative diseases such as arterial hypertension and diabetes mellitus to constitute the physiopathological basis of cardio- and cerebrovascular accidents.
- diabetes
- stroke
- cerebrovascular disease
- atherosclerosis
1. Vascular Complications of Diabetes Mellitus
Type 2D diabetes (T2D) and atherosclerosis are, from the point of view of cardio- and cerebrovascular risk, two complementary diseases. Beyond shared aspects such as inflammation and oxidative stress, there are multiple molecular mechanisms by which they feed off each other: chronic hyperglycemia and advanced glycosylation end-products (AGE) promote ‘accelerated atherosclerosis’ through the induction of endothelial damage and cellular dysfunction.
These diseases impact the vascular system and, therefore, the risk of developing cardio- and cerebrovascular events is now evident, but the observation of this significant correlation has its roots in past decades.
There have been several findings of experiences in which this correlation seems to be evident in scientific literature. For example, in 1969, Heyden et al. reported retrospective case-control studies and prospective studies from Los Angeles and Framingham, in which it was apparent that hypertension and DM were risk factors for cerebrovascular diseases and that the early treatment of DM could be essential to prevent or delay the onset of stroke [34][1].
Over the years, it has become clear that the role of T2D is decisive in triggering cardio and cerebrovascular events: in 1977, Królewski et al. made a retrospective analysis through which observed, in diabetic patients, a mortality rate 1.3 times higher than in non-diabetic subjects and noticed that the causes of death were mainly coronary heart disease and cerebrovascular disease. Furthermore, the risk of death for cardiovascular diseases was greater in patients with early onset of diabetes, as evidence that the duration of the disease and its pathophysiological mechanisms are connected with vascular disorders. Moreover, these observations were possible because of the discovery of insulin and its use for the treatment of diabetes: this drug prolonged the life expectancy of diabetic patients by reducing acute complications, such as acidotic coma, and made it possible to observe vascular complications over time [35][2].
Although diabetes was often considered a risk factor for myocardial infarction or ischemic stroke in the context of an atherogenic profile, several scientific works have shown the role of T2D as an independent risk factor for cardio and cerebrovascular diseases. For example, the Honolulu Heart Program, a prospective study of cardiovascular disease, assessed the risk of stroke considering two cohorts of patients (diabetic patients vs. non-diabetic subjects without coronary heart disease and history of stroke at the beginning of the study). The relative risk of having an ischemic thromboembolic stroke for diabetic patients compared with non-diabetic subjects was 2.0 (95% confidence limit, 1.4 to 3.0), and it was observed that tight control of other atherogenic conditions (i.for example., hypertension, hypercholesterolemia, sedentary lifestyle) did not decrease the effect of diabetes in causing the stroke. Therefore, diabetes mellitus appears to be an additional independent risk factor for ischemic stroke (no correlation between diabetes and hemorrhagic stroke was observed) [36][3].
The significant impact of vascular diseases in diabetic patients has become increasingly important: it is now clear that diabetic people have an increased risk of coronary heart disease, peripheral arterial occlusive disease, and death for cardiovascular causes compared with non-diabetic subjects. The Multiple Risk Factor Intervention Trial, in 1993, showed this aspect more sharply: the incidence of cardiovascular diseases and CHD (coronary heart disease) mortality increases in both diabetic and non-diabetic cohorts of patients as cholesterol levels rise, but the mortality rate among people with diabetes is four or five times higher than non-diabetics at the same cholesterol levels [37][4].
The correlation between the duration of diabetes and the risk of cerebrovascular disease has become progressively evident. For example, it has been found that among patients with stroke younger than 55 years, those with diabetes mellitus had a risk more than 10-fold [38][5].
To confirm the above, the experience of the Baltimore-Washington Cooperative Young Stroke Study is crucial: were enrolled 296 cases of ischemic stroke among young adults from 18 to 44 years and, through this analysis, the study showed that diabetes increased the odds ratio for stroke, from 3.3 for black women to 23.1 for white men evidence that, in addition to gender differences, diabetes plays a predominant role in the risk profile for stroke [39][6].
Patients with diabetes of all age groups have a probability at least twice of stroke compared with non-diabetic subjects. In addition to the duration of diabetes, there is another factor to evaluate the prognosis of diabetic patients with stroke: they have an elevated risk of a future ischemic event. The close connection between DM and ischemic stroke, therefore, is not only due to the comorbidities of diabetic patients who have a cerebrovascular disease (i.for example., hypertension, hypercholesterolemia) but also to diabetes-specific properties involving small penetrating arteries: people with diabetes have a significant probability of suffering a lacunar stroke or small vessel disease [40][7].
2. Molecular Pathology of Vascular Damage in Diabetes Mellitus and Atherosclerosis
The correlation between diabetes mellitus and ischemic stroke has been found by several authors and is a common experience in clinical practice. In agreement with previous studies, Matz et al. discovered a high prevalence of glucose metabolism disorders in patients with ischemic stroke, including new-onset diabetes mellitus. While diabetes is a known risk factor for the occurrence of ischemic stroke, it remains to be understood whether asymptomatic hyperglycemia has a potential role in causing a cerebrovascular event [41,42][8][9].
The frequency of T2D is three times higher in patients with ischemic stroke than in controls [43][10].
Indeed, the risk of stroke increases from 150% to 400% in diabetics, and the extent of glycemic dyscontrol correlates directly with the risk of acute cerebrovascular accidents [41,44][8][11].
The altered metabolic framework that characterizes diabetes mellitus influences vascular changes. The most characteristic abnormalities certainly include chronic hyperglycemia, insulin resistance, and dyslipidemia: these factors can promote the atherosclerotic structure and induce cellular dysfunction at various levels (like endothelial dysfunction, smooth muscle cell alterations, platelet abnormalities, and coagulation alterations).
Concerning endothelial dysfunction, it should be remembered that endothelial cells represent the inner lining of the vascular lumen and perform, in addition to the mechanical function of coating, an endocrine role through the production of biologically active substances that regulate various functions: nitric oxide, prostaglandins, endothelin-1, angiotensin II, and other reactive oxygen species. Nitric oxide dilates the vessels, inhibits platelet activation, reduces the proliferation of smooth muscle cells, and reduces the process of diapedesis of leukocytes. The combination of these phenomena aims to limit the atherosclerotic pattern, ensuring the vascular system’s integrity [45,46,47][12][13][14].
Several studies have shown that T2D alters endothelial function through a cascade of molecular events that precede atherosclerotic plaque formation but promote its appearance. Constant hyperglycemia inhibits the enzyme eNOS (endothelial nitric oxide synthase) and thus cause the reduction of nitric oxide and the stimulation of the production of reactive oxygen species, including superoxide anion (O2−) [48][15]; superoxide anion neutralizes nitric oxide by producing the toxic ion peroxynitrite, which uncouples the eNOS enzyme by oxidizing its cofactor, tetrahydrobiopterin [49][16].
Another mechanism that limits the production of nitric oxide is related to insulin resistance: the reduced action of insulin on adipocytes leads to an increased release of free fatty acids from adipose tissue [50][17] with consequent activation of the pathway of protein kinase C, then inhibition of phosphatidylinositol-3 (PI-3) kinase and increase of reactive oxygen species at the expense of nitric oxide [51][18].
While, from what has been said above, T2D reduces the production of mediators of vasodilation, it is also true that in diabetic patients, the production of vasoconstrictive substances increases, including, crucially, endothelin-1.
Endothelin-1 has a dual effect: it promotes vasoconstriction through its action on smooth muscle and activates renal salt and water retention, resulting in activation of the renin-angiotensin-aldosterone system and thus smooth muscle hypertrophy [52][19].
The concentration of endothelin-1, among other things, increases in response to the insulin-mediated effects of increased gene expression and receptor synthesis and as a consequence of the increased presence of glycosylation products, so its vaso-active effects in diabetes are multiple and complex [53][20].
Moreover, endothelial cells regulate the cell transit through the vessel wall producing chemotactic adhesion molecules: monocytes, once reaching the subendothelial space, phagocytize oxidized LDL and become foamy cells, the initial substrate of atherosclerotic lesions [23][21].
Advanced glycosylation end-products (AGEs) also amplify endothelial cell damage: high serum glucose levels produce a process of glycation and glycosylation of proteins at the extracellular level, leading to their accumulation. These molecular products cause the increased expression of adhesion molecules on endothelial cells, promote the migration of monocytes/macrophages towards the forming atherosclerotic plaque and promote the release of inflammatory cytokines by histiocytes. They also contribute to changes in the extracellular matrix and are a risk factor for plaque rupture [54,55][22][23] (Figure 1).
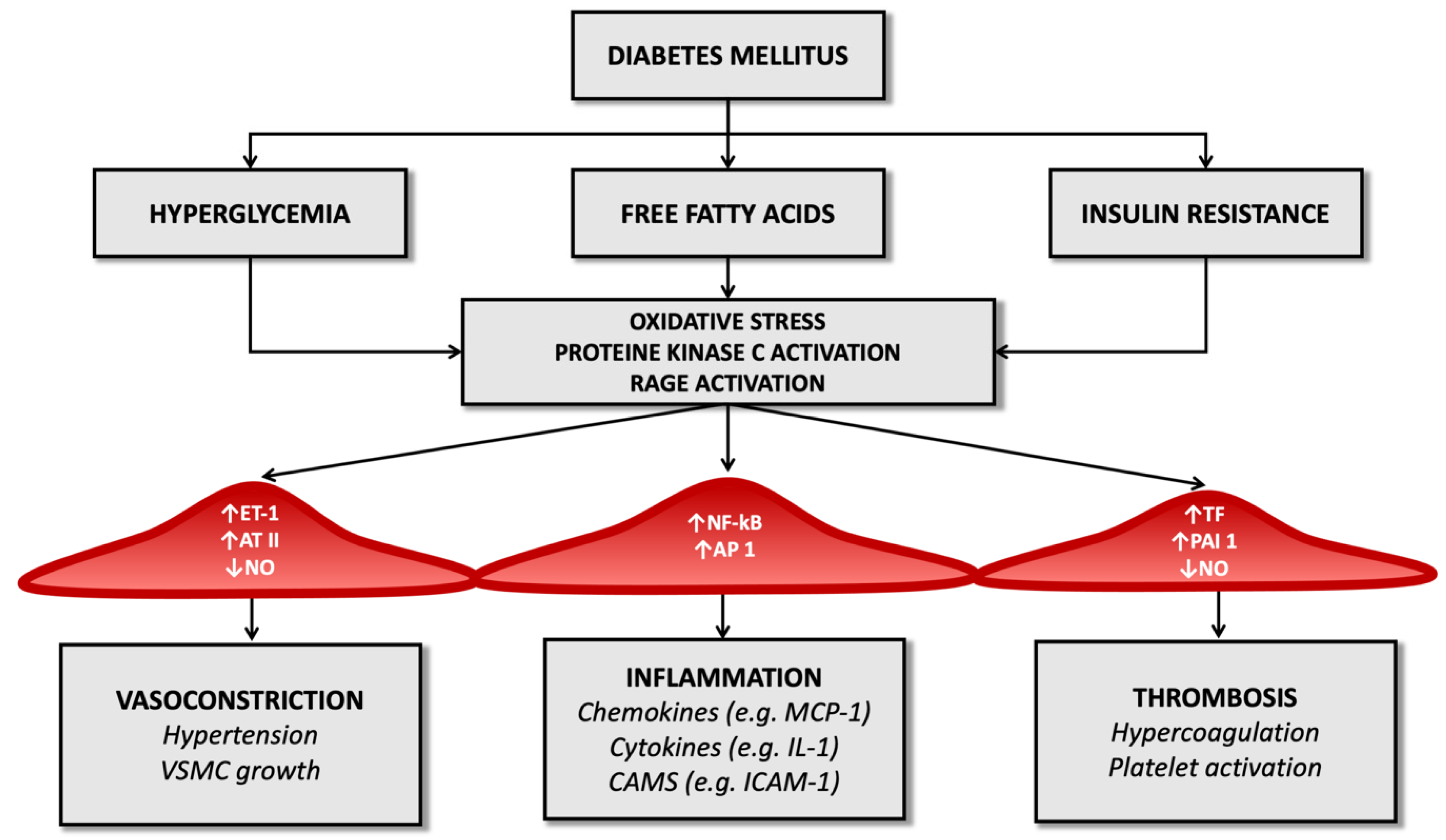
Figure 1.
Diabetes’ mechanisms of vascular damage.
In the diabetic patient, therefore, all these molecular pathways (hyperglycemia, increased oxidative stress, and activation of receptors for advanced products of glycosylation) promote the expression of the gene for the nuclear factor kappa-light-chain-enhancer of activated B cells (Nf-kB): this leads to increased production of inflammatory mediators (such as interleukin-1) and adhesion molecules for leukocytes that favor atherogenesis [17,56,57][24][25][26].
Thus, endothelial dysfunction represents a significant factor in the pathogenesis of ischemic stroke and is a shared effect of diabetes mellitus and atherosclerosis, the ‘accelerator pedal’ on which both these diseases press.
The reduced bioavailability of nitric oxide and the prevalence of molecules mediating vasoconstriction in people with diabetes results in altered smooth muscle cell function: infusion of angiotensin II or endothelin-1, in fact, in these patients, results in a lower vasoconstriction effect than in healthy controls [58,59,60][27][28][29].
Among other things, diabetic patients have autonomic dysfunction that modifies peripheral vascular resistance through mechanisms that are not yet fully known and this, together with the above, constitutes an essential element of vascular damage [61][30].
The altered metabolic pattern generated by diabetes also results in structural and functional alterations in smooth muscle cells. In non-diabetic subjects, during the atherogenic process, muscle cells migrate from the intermediate layer of the vascular wall to the forming atherosclerotic plaque, replicating and contributing to the extracellular matrix’s constitution. In people with T2D, an increased migration capacity of smooth muscle cells has been demonstrated in vitro. Still, these cells are reduced in complicated atherosclerotic plaques [62][31] since hyperglycemia determines modifications on the oxidation process of low-density lipoprotein (LDL) that favor their apoptosis [63][32].
The progression of atherosclerosis and the risk of plaque rupture in diabetic patients is also linked to platelet dysfunction generated by the disease: the concentration of glucose inside platelets, like endothelial cells, does not depend on the action of insulin but the extracellular concentration of glucose. The increased intracellular availability of glucose causes activation of protein kinase C, reduced nitric oxide production, and increased reactive oxygen species, as mentioned above about endothelial cells [64][33].
The result is impaired platelet function, altered homeostasis of intracellular calcium, and dysregulation of the thromboxane synthesis process. In addition, in diabetic patients, increased surface expression of glycoprotein Ib (which mediates interaction with von Willebrand factor) and glycoprotein IIb/IIIa, which regulates interaction with fibrin and leads to a significant increase in potential thrombotic risk [65][34].
Platelet alteration is only one aspect of the alterations inherent in the coagulation process. For example, several studies have observed that in diabetic patients, there is reduced fibrinolytic activity due to the high concentration of the tissue plasminogen activator inhibitor type 1 both in atheromatous plaques and in arteries not affected by the atheromatous process [66][35].
Diabetes also determines an imbalance between factors that regulate the hemostatic balance, favoring the increased availability of tissue factor and factor VII of the coagulation cascade (which have a potent procoagulant activity) and determining a reduction in serum levels of anticoagulant factors such as protein C and antithrombin III (Figure 1). It seems that these alterations depend directly on the condition of hyperglycemia that characterizes T2D and partly on the cleavage products of proinsulin in the process of transformation to insulin [67][36].
Chronic inflammation is another determinant of thrombotic risk and is a common feature of atherosclerosis and diabetes mellitus. Increased inflammasome activity and high levels of nucleotide-binding oligomerization domain-like receptor 3 (NLRP3) have been documented in diabetic patients, together with increased serum levels of pro-inflammatory cytokines such as interleukin-1 beta and interleukin-18. One of the common aspects of diabetes and atherosclerosis in the pattern of inflammation is neutrophil extracellular trap activation, or NETosis, a particular type of cell death in macrophages through which chromatin is released into the extracellular space to trap and kill bacteria. This mechanism is typical of chronic inflammatory conditions and infections, and it has been observed that it can be promoted by hyperglycemia [68][37].
Additionally, in animal models, NETosis has been shown to promote atherosclerosis so that, through anti-diabetic therapy, it is possible to limit the atherosclerotic burden by defining the process of atherosclerotic plaque genesis.
In addition to the already known mechanisms by which T2D promotes inflammation, recently, the high-mobility group box 1 protein (HMGB1), non-histone proteins that act as an alarm for the immune system to initiate tissue repair and host defence processes, have been increasingly studied. It appears that during the acute phase of stroke, these proteins migrate towards the extracellular space and are bound by Toll-like receptors (TLR-2, TLR-4) and receptors for advanced glycosylation end-products (RAGE) that activate the transcription factor Nf-kB and the synthesis of inflammatory cytokines that worsen outcome and prognosis [69][38].
According to [60][29], in diabetic patients, the excess presence of advanced glycosylation end-products (AGEs) and their receptors (RAGEs) is an unfavorable element, so the possibility of limiting the expression of HMGB1, especially in diabetics, is being investigated to limit the inflammatory pattern of ischemic stroke and to improve the prognosis of these patients.
Altogether, diabetes mellitus is a true generator of thrombotic risk and, therefore, a promoter of ischemic stroke: on the one hand, it activates molecular damage mechanisms leading to endothelial dysfunction and the progression of the atherosclerotic process, and on the other, it increases the thrombogenic risk through the induction of platelet dysfunction and the dysregulation of the coagulation cascade.
3. Epidemiology of Ischemic Stroke in T2DM Patients
Type 2 diabetes mellitus (T2DM) is one of the most common chronic pathologies, and in 2015 nearly 400 million subjects in the world were considered diabetic; the prevalence of this disease is supposed to increase up to 640 million people in the next 20 years [70][39].
Stroke affects more than 500,000 persons every year in the United States and represents one of the most significant reasons for decease in the Western part of the world [71][40]; in addition, ischemic stroke is the second most frequent complication of T2DM after coronary artery disease (CAD) [72][41], and the second source of death after cancer in diabetic subjects [73][42].
The Framingham Study showed that the incidence of ischemic stroke in diabetic patients was 2.5–3.5 times increased compared to the control group [74][43]. Similar results have been observed in multiple research [75,76][44][45].
While cardioembolic stroke is more common in non-diabetic subjects, diabetes is correlated to cerebral ischemia caused by atherosclerosis [77][46]; the endothelial dysfunction can easily explain this due to the persistent hyperglycemia and by the concomitant presence of other risk factors for atherosclerosis, such as hypertension and hyperlipidemia.
In an observational study, Mulnier et al. reported a rate of stroke of 11.9 per 1000 persons per year in subjects affected by DM, in the healthy group, this rate was 5.5 per 1000 person-year; the highest hazard ratio correlated with ischemic stroke was noticed in the 35–54 year group, in addition, the risk was significantly higher in women than in men [78][47]. As observed by Selvin et al., there is a correlation between the levels of glycated hemoglobin (HbA1c) and the chance of cerebral ischemia [79][48]. Furthermore, proteinuria (>300 mg/dL) has been associated with an increased risk of ischemic stroke, even if it does not seem to influence the prognosis [80][49].
T2DM is rarely an isolated condition; in the majority of the cases, it is associated with other well-known risk factors for ischemic stroke, such as dyslipidemia and hypertension. Consequently, it is necessary to operate on these pathologies to reduce the chance of cerebral ischemia in diabetic subjects.
Kearney et al. performed a meta-analysis of 14 randomized trials including more than 18,000 subjects affected by diabetes mellitus; statin therapy led to a significant reduction of the risk of ischemic stroke; this reduction was more marked in diabetic patients than in the control group [81][50].
As far as it concerns hypertension, the UK Prospective Diabetes Study (UKPDS) showed that the reduction of 10 mmHg in systolic blood pressure resulted in a more than 40% decrease in stroke incidence [82][51]. Furthermore, treatment with indapamide plus perindopril was associated with a 38% diminution of the stroke risk in patients affected by diabetes mellitus, as reported by the Perindopril Protection Against Recurrent Stroke Study (PROGRESS) [83][52].
Moreover, Lichtman et al. reported diabetes as an independent risk factor for cerebral ischemia in the first six months after acute coronary syndrome [84][53]; the same finding was observed after coronary artery bypass grafting [85][54].
Diabetes mellitus is a common comorbidity in subjects affected by an ischemic stroke. Up to 20% of stroke patients have diabetes [86,87,88][55][56][57]; furthermore, Gray et al. reported a new diagnosis of diabetes by 12 weeks after the stroke in nearly 20% of the subjects [89][58].
Multiple studies reported the significant association between DM and lacunar stroke [90][59]. This finding is quite plausible because hyperglycemia, together with hypertension, plays a crucial role in the process of lipohyalinosis, which is the leading cause of small vessel disease. Nevertheless, some studies did not find this association [91,92][60][61].
References
- Heyden, S.; Gerber, C.J. Atherosclerotic Cerebrovascular Disease —Its Nature and Management. Am. J. Med. 1969, 46, 763–773.
- Królewski, A.S.; Czyzyk, A.; Janeczko, D.; Kopczyński, J. Mortality from Cardiovascular Diseases among Diabetics. Diabetologia 1977, 13, 345–350.
- Abbott, R.D.; Donahue, R.P.; MacMahon, S.W.; Reed, D.M.; Yano, K. Diabetes and the Risk of Stroke. JAMA 1987, 257, 949–952.
- Stamler, J.; Vaccaro, O.; Neaton, J.D.; Wentworth, D. Diabetes, Other Risk Factors, and 12-Yr Cardiovascular Mortality for Men Screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993, 16, 434–444.
- You, R.X.; McNeil, J.J.; O’Malley, H.M.; Davis, S.M.; Thrift, A.G.; Donnan, G.A. Risk Factors for Stroke Due to Cerebral Infarction in Young Adults. Stroke 1997, 28, 1913–1918.
- Rohr, J.; Kittner, S.; Feeser, B.; Hebel, J.R.; Whyte, M.G.; Weinstein, A.; Kanarak, N.; Buchholz, D.; Earley, C.; Johnson, C.; et al. Traditional Risk Factors and Ischemic Stroke in Young Adults: The Baltimore-Washington Cooperative Young Stroke Study. Arch. Neurol. 1996, 53, 603–607.
- Hill, M.D. Stroke and Diabetes Mellitus. Handb. Clin. Neurol. 2014, 126, 167–174.
- Folsom, A.R.; Rasmussen, M.L.; Chambless, L.E.; Howard, G.; Cooper, L.S.; Schmidt, M.I.; Heiss, G. Prospective Associations of Fasting Insulin, Body Fat Distribution, and Diabetes with Risk of Ischemic Stroke. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Diabetes Care 1999, 22, 1077–1083.
- Goldstein, L.B.; Adams, R.; Becker, K.; Furberg, C.D.; Gorelick, P.B.; Hademenos, G.; Hill, M.; Howard, G.; Howard, V.J.; Jacobs, B.; et al. Primary Prevention of Ischemic Stroke: A Statement for Healthcare Professionals from the Stroke Council of the American Heart Association. Circulation 2001, 103, 163–182.
- Himmelmann, A.; Hansson, L.; Svensson, A.; Harmsen, P.; Holmgren, C.; Svanborg, A. Predictors of Stroke in the Elderly. Acta Med. Scand. 1988, 224, 439–443.
- Kuusisto, J.; Mykkänen, L.; Pyörälä, K.; Laakso, M. Non-Insulin-Dependent Diabetes and Its Metabolic Control Are Important Predictors of Stroke in Elderly Subjects. Stroke 1994, 25, 1157–1164.
- Verma, S.; Anderson, T.J. The Ten Most Commonly Asked Questions about Endothelial Function in Cardiology. Cardiol. Rev. 2001, 9, 250–252.
- Sarkar, R.; Meinberg, E.G.; Stanley, J.C.; Gordon, D.; Webb, R.C. Nitric Oxide Reversibly Inhibits the Migration of Cultured Vascular Smooth Muscle Cells. Circ. Res. 1996, 78, 225–230.
- Kubes, P.; Suzuki, M.; Granger, D.N. Nitric Oxide: An Endogenous Modulator of Leukocyte Adhesion. Proc. Natl. Acad. Sci. USA 1991, 88, 4651–4655.
- De Vriese, A.S.; Verbeuren, T.J.; Van de Voorde, J.; Lameire, N.H.; Vanhoutte, P.M. Endothelial Dysfunction in Diabetes. Endothel. Dysfunct. Diabetes. Br. J. Pharmacol. 2000, 130, 963–974.
- Milstien, S.; Katusic, Z. Oxidation of Tetrahydrobiopterin by Peroxynitrite: Implications for Vascular Endothelial Function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684.
- Hennes, M.M.; O’Shaughnessy, I.M.; Kelly, T.M.; LaBelle, P.; Egan, B.M.; Kissebah, A.H. Insulin-Resistant Lipolysis in Abdominally Obese Hypertensive Individuals. Hypertension 1996, 28, 120–126.
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High Glucose Level and Free Fatty Acid Stimulate Reactive Oxygen Species Production through Protein Kinase C--Dependent Activation of NAD(P)H Oxidase in Cultured Vascular Cells. Diabetes 2000, 49, 1939–1945.
- Hopfner, R.L.; Gopalakrishnan, V. Endothelin: Emerging Role in Diabetic Vascular Complications. Diabetologia 1999, 42, 1383–1394.
- Quehenberger, P.; Bierhaus, A.; Fasching, P.; Muellner, C.; Klevesath, M.; Hong, M.; Stier, G.; Sattler, M.; Schleicher, E.; Speiser, W.; et al. Endothelin 1 Transcription Is Controlled by Nuclear Factor-KappaB in AGE-Stimulated Cultured Endothelial Cells. Diabetes 2000, 49, 1561–1570.
- Libby, P. Inflammation in Atherosclerosis. Nature 2002, 420, 868–874.
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39.
- Khan, M.I.; Pichna, B.A.; Shi, Y.; Bowes, A.J.; Werstuck, G.H. Evidence Supporting a Role for Endoplasmic Reticulum Stress in the Development of Atherosclerosis in a Hyperglycaemic Mouse Model. Antioxid. Redox Signal. 2009, 11, 2289–2298.
- Schmidt, A.M. Highlighting Diabetes Mellitus: The Epidemic Continues. Arter. Thromb. Vasc. Biol. 2018, 38, e1–e8.
- Rösen, P.; Nawroth, P.P.; King, G.; Möller, W.; Tritschler, H.J.; Packer, L. The Role of Oxidative Stress in the Onset and Progression of Diabetes and Its Complications: A Summary of a Congress Series Sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes/Metab. Res. Rev. 2001, 17, 189–212.
- Zeiher, A.M.; Fisslthaler, B.; Schray-Utz, B.; Busse, R. Nitric Oxide Modulates the Expression of Monocyte Chemoattractant Protein 1 in Cultured Human Endothelial Cells. Circ. Res. 1995, 76, 980–986.
- Christlieb, A.R.; Janka, H.U.; Kraus, B.; Gleason, R.E.; Icasas-Cabral, E.A.; Aiello, L.M.; Cabral, B.V.; Solano, A. Vascular Reactivity to Angiotensin II and to Norepinephrine in Diabetic Subjects. Diabetes 1976, 25, 268–274.
- Nugent, A.G.; McGurk, C.; Hayes, J.R.; Johnston, G.D. Impaired Vasoconstriction to Endothelin 1 in Patients with NIDDM. Diabetes 1996, 45, 105–107.
- Tesfamariam, B.; Cohen, R.A. Enhanced Adrenergic Neurotransmission in Diabetic Rabbit Carotid Artery. Cardiovasc. Res. 1995, 29, 549–554.
- McDaid, E.A.; Monaghan, B.; Parker, A.I.; Hayes, J.R.; Allen, J.A. Peripheral Autonomic Impairment in Patients Newly Diagnosed with Type II Diabetes. Diabetes Care 1994, 17, 1422–1427.
- Fukumoto, H.; Naito, Z.; Asano, G.; Aramaki, T. Immunohistochemical and Morphometric Evaluations of Coronary Atherosclerotic Plaques Associated with Myocardial Infarction and Diabetes Mellitus. J. Atheroscler. Thromb. 1998, 5, 29–35.
- Taguchi, S.; Oinuma, T.; Yamada, T. A Comparative Study of Cultured Smooth Muscle Cell Proliferation and Injury, Utilizing Glycated Low Density Lipoproteins with Slight Oxidation, Auto-Oxidation, or Extensive Oxidation. J. Atheroscler. Thromb. 2000, 7, 132–137.
- Assert, R.; Scherk, G.; Bumbure, A.; Pirags, V.; Schatz, H.; Pfeiffer, A.F. Regulation of Protein Kinase C by Short Term Hyperglycemia in Human Platelets in Vivo and in Vitro. Diabetologia 2001, 44, 188–195.
- Vinik, A.I.; Erbas, T.; Park, T.S.; Nolan, R.; Pittenger, G.L. Platelet Dysfunction in Type 2 Diabetes. Diabetes Care 2001, 24, 1476–1485.
- Carr, M.E. Diabetes Mellitus: A Hypercoagulable State. J. Diabetes Complicat. 2001, 15, 44–54.
- Nordt, T.K.; Bode, C. Impaired Endogenous Fibrinolysis in Diabetes Mellitus: Mechanisms and Therapeutic Approaches. Semin. Thromb. Hemost. 2000, 26, 495–501.
- Joshi, M.B.; Lad, A.; Prasad, A.S.B.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High Glucose Modulates IL-6 Mediated Immune Homeostasis through Impeding Neutrophil Extracellular Trap Formation. FEBS Lett. 2013, 587, 2241–2246.
- Ye, Y.; Zeng, Z.; Jin, T.; Zhang, H.; Xiong, X.; Gu, L. The Role of High Mobility Group Box 1 in Ischemic Stroke. Front. Cell. Neurosci. 2019, 13, 127.
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global Estimates for the Prevalence of Diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50.
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492.
- Bertoni, A.G.; Krop, J.S.; Anderson, G.F.; Brancati, F.L. Diabetes-Related Morbidity and Mortality in a National Sample of U.S. Elders. Diabetes Care 2002, 25, 471–475.
- Katakura, M.; Naka, M.; Kondo, T.; Nishii, N.; Komatsu, M.; Sato, Y.; Yamauchi, K.; Hiramatsu, K.; Ikeda, M.; Aizawa, T.; et al. Prospective Analysis of Mortality, Morbidity, and Risk Factors in Elderly Diabetic Subjects: Nagano Study. Diabetes Care 2003, 26, 638–644.
- Kannel, W.B.; McGee, D.L. Diabetes and Cardiovascular Disease. The Framingham Study. JAMA 1979, 241, 2035–2038.
- Kissela, B.M.; Khoury, J.; Kleindorfer, D.; Woo, D.; Schneider, A.; Alwell, K.; Miller, R.; Ewing, I.; Moomaw, C.J.; Szaflarski, J.P.; et al. Epidemiology of Ischemic Stroke in Patients with Diabetes: The Greater Cincinnati/Northern Kentucky Stroke Study. Diabetes Care 2005, 28, 355–359.
- Almdal, T.; Scharling, H.; Jensen, J.S.; Vestergaard, H. The Independent Effect of Type 2 Diabetes Mellitus on Ischemic Heart Disease, Stroke, and Death: A Population-Based Study of 13,000 Men and Women with 20 Years of Follow-Up. Arch. Intern. Med. 2004, 164, 1422–1426.
- Tuttolomondo, A.; Pinto, A.; Salemi, G.; Di Raimondo, D.; Di Sciacca, R.; Fernandez, P.; Ragonese, P.; Savettieri, G.; Licata, G. Diabetic and Non-Diabetic Subjects with Ischemic Stroke: Differences, Subtype Distribution and Outcome. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 152–157.
- Mulnier, H.E.; Seaman, H.E.; Raleigh, V.S.; Soedamah-Muthu, S.S.; Colhoun, H.M.; Lawrenson, R.A.; De Vries, C.S. Risk of Stroke in People with Type 2 Diabetes in the UK: A Study Using the General Practice Research Database. Diabetologia 2006, 49, 2859–2865.
- Selvin, E.; Coresh, J.; Shahar, E.; Zhang, L.; Steffes, M.; Sharrett, A.R. Glycaemia (Haemoglobin A1c) and Incident Ischaemic Stroke: The Atherosclerosis Risk in Communities (ARIC) Study. Lancet Neurol. 2005, 4, 821–826.
- Guerrero-Romero, F.; Rodríguez-Morán, M. Proteinuria Is an Independent Risk Factor for Ischemic Stroke in Non-Insulin-Dependent Diabetes Mellitus. Stroke 1999, 30, 1787–1791.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney, P.M.; Blackwell, L.; Collins, R.; Keech, A.; Simes, J.; Peto, R.; Armitage, J.; Baigent, C. Efficacy of Cholesterol-Lowering Therapy in 18,686 People with Diabetes in 14 Randomised Trials of Statins: A Meta-Analysis. Lancet 2008, 371, 117–125.
- UK Prospective Diabetes Study Group. Tight Blood Pressure Control and Risk of Macrovascular and Microvascular Complications in Type 2 Diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998, 317, 703–713.
- PROGRESS Collaborative Group Randomised Trial of a Perindopril-Based Blood-Pressure-Lowering Regimen among 6105 Individuals with Previous Stroke or Transient Ischaemic Attack. Lancet 2001, 358, 1033–1041.
- Lichtman, J.H.; Krumholz, H.M.; Wang, Y.; Radford, M.J.; Brass, L.M. Risk and Predictors of Stroke after Myocardial Infarction among the Elderly: Results from the Cooperative Cardiovascular Project. Circulation 2002, 105, 1082–1087.
- D’Ancona, G.; de Ibarra, J.I.S.; Baillot, R.; Mathieu, P.; Doyle, D.; Metras, J.; Desaulniers, D.; Dagenais, F. Determinants of Stroke after Coronary Artery Bypass Grafting. Eur. J. Cardio-Thorac. Surg. 2003, 24, 552–556.
- Anderson, C.S.; Carter, K.N.; Hackett, M.L.; Feigin, V.; Barber, P.A.; Broad, J.B.; Bonita, R.; Auckland Regional Community Stroke (ARCOS) Study Group. Trends in Stroke Incidence in Auckland, New Zealand, during 1981 to 2003. Stroke 2005, 36, 2087–2093.
- Benatru, I.; Rouaud, O.; Durier, J.; Contegal, F.; Couvreur, G.; Bejot, Y.; Osseby, G.V.; Ben Salem, D.; Ricolfi, F.; Moreau, T.; et al. Stable Stroke Incidence Rates but Improved Case-Fatality in Dijon, France, from 1985 to 2004. Stroke 2006, 37, 1674–1679.
- Rothwell, P.M.; Coull, A.J.; Giles, M.F.; Howard, S.C.; Silver, L.E.; Bull, L.M.; Gutnikov, S.A.; Edwards, P.; Mant, D.; Sackley, C.M.; et al. Change in Stroke Incidence, Mortality, Case-Fatality, Severity, and Risk Factors in Oxfordshire, UK from 1981 to 2004 (Oxford Vascular Study). Lancet 2004, 363, 1925–1933.
- Gray, C.S.; Scott, J.F.; French, J.M.; Alberti, K.G.M.M.; O’Connell, J.E. Prevalence and Prediction of Unrecognised Diabetes Mellitus and Impaired Glucose Tolerance Following Acute Stroke. Age Ageing 2004, 33, 71–77.
- Karapanayiotides, T.; Piechowski-Jozwiak, B.; van Melle, G.; Bogousslavsky, J.; Devuyst, G. Stroke Patterns, Etiology, and Prognosis in Patients with Diabetes Mellitus. Neurology 2004, 62, 1558–1562.
- Jackson, C.; Sudlow, C. Are Lacunar Strokes Really Different? A Systematic Review of Differences in Risk Factor Profiles between Lacunar and Nonlacunar Infarcts. Stroke 2005, 36, 891–901.
- Schulz, U.; Rothwell, P. Differences in Vascular Risk Factors Between Etiological Subtypes of Ischemic Stroke. Stroke 2003, 34, 2050–2059.
More