Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Lucio Cocco and Version 2 by Catherine Yang.
Renal failure is a worldwide disease with a continuously increasing prevalence and involving a rising need for long-term treatment, mainly by haemodialysis. Arteriovenous fistula (AVF) is the favourite type of vascular access for haemodialysis; however, the lasting success of this therapy depends on its maturation, which is directly influenced by many concomitant processes such as vein wall thickening or inflammation. Understanding the molecular mechanisms that drive AVF maturation and failure can highlight new or combinatorial drugs for more personalized therapy.
- AVF fistula
- PI3K
- AKT mTOR
1. PI3K/AKT/mTOR Axis
Phosphoinositide 3-kinases are enzymes which catalyse the phosphorylation of one or more inositol phospholipids on the 3-position of the inositol ring [1][34]: in mammalian cells there are eight PI3K isoforms that can be divided into three classes based on common substrate specificity, structural similarities and an analogous mechanism of activation.
Class I PI3Ks are well-characterized enzymes that respond to signals received through cell surface receptors such as activated tyrosine kinase receptors (RTKs), G protein-coupled receptors (GPCRs) and monomeric small GTPases [2][3][35,36]. Class I PI3Ks are heterodimers made by a regulatory subunit and a catalytic subunit [4][37]. Once stimulated, the receptor becomes activated and its intracellular portion is able to be bound by a regulatory subunit, which in turn activates the catalytic subunits that phosphorylate PtdIns(4,5)P2 into PtdIns(3,4,5)P3 at the lipid membranes [5][38]. Among the different isoforms, PI3Kα and PI3Kβ are relatively ubiquitously expressed, while the PI3Kγ and PI3Kδ isoforms are enriched mainly in hematopoietic cells, such as leukocytes [3][6][36,39].
Class II PI3Ks have distinguishing, non-overlapping functions and are involved mainly in endocytosis, membrane dynamics and trafficking [7][40]. Class II PI3Ks are monomers present in three isoforms (PI3K-C2α,-C2β and -C2γ): in contrast to Class I PI3Ks, they are not stimulated by a cell surface receptor, but are activated by internal cell signalling [8][41]. PI3KC2α and PI3KC2β are expressed ubiquitously, while PI3KC2γ is present mainly in the liver [9][42].
Class III PI3K is represented by a single isoform known as vacuolar protein sorting 34 (Vps34) occupied in controlling vesicular endosome-lysosome sorting, which might induce pathogen killing, antigen processing and immune cell survival [2][10][35,43].
The three classes generate diverse lipid products, and therefore recruit different downstream effectors. Notably for this review, Class I, but not Class II, phosphorylates PtdIns(4,5)P2 into PtdIns(3,4,5)P3, which in turn drives the localisation of protein kinase B (PKB, also known as AKT) to the plasma membrane together with its upstream regulatory kinases. Co-localisation at the plasma membrane essentially increases the effective concentration of AKT and its kinases, thereby increasing the likelihood of interaction. In addition, interaction of 3-phosphoinositide-binding pleckstrin homology (PH) domain of AKT with PtdIns(3,4,5)P3 also induces a conformational switch which enables AKT phosphorylation by its upstream activators. AKT is a serine/threonine kinase with a multitude of substrates and targets; this in part explains the relatively wide spectrum of effects of PtdIns(3,4,5)P3 signalling on downstream pathways.
Another PI3K effector can be mTOR. mTOR exists in two multi-protein complexes, mTORC1 and mTORC2, with distinct subunit composition, substrate selectivity and activity and sensitivity to sirolimus (rapamycin). A well-established substrate of mTORC2 is AKT, phosphorylated in its Ser473, in addition to protein kinase C (PKC) isoforms and serum- and glucocorticoid-regulated kinases (SGKs) [1][34]. mTORC1 phosphorylates several substrates that induce anabolic metabolism to sustain growth and proliferation [1][34]. mTORC1 action can be induced by proliferating stimuli via PI3K/AKT, RAS/ERK and other signalling cascades [11][44].
Overall, the PI3K/AKT/mTOR axis transmits important signals that regulate a variety of physiological processes in virtually all tissue types studied to date (which have been extensively described in literature) such as proliferation, apoptosis, inflammation cellular migration and immune response, and its dysregulation is associated with development of various diseases including cancer, diabetes and autoimmunity: Here we describe highlights of the PI3K/Akt7mTORC1 axis in arteriovenous fistula patency.
2. PI3K/Akt/mTOR Pathway in AVF
Unfortunately, AV fistulas fail to mature in 30–50% of cases (early failure) and a mature correct AV fistula fails in one year (late failure) in 40% of cases, reflecting a dramatic clinical outcome and the urgent need for more a accurate understanding of the mechanisms that impair the fistula. During AVF vein remodelling, a complex scenario takes place involving interconnected processes such as inflammation, VSMC proliferation and consequently neointima hyperplasia, extracellular matrix deposition and adventitia remodelling: in all these functions PI3K/AKT/mTOR signalling pathways can be implicated with different and even contrasting outcomes.
VSMCs dedifferentiation can be induced by growth factors and inflammatory cytokines locally released after vascular injury [12][16]: among these, platelet-derived growth factor (PDGF) is one of the most powerful mitogens that can induce a “phenotypic switch” from a contractile to a proliferative state, an essential step for neointima hyperplasia. PDGF-induced VSMC proliferation [13][14][45,46] is mediated by a signalling cascade that includes PI3K/AKT, mTOR/p70S6kinase and MEK1/ERK signalling downstream of the insulin receptor stimulus. Moreover, by interfering with dysregulated insulin receptor signalling, Zhao et al., postulated new therapeutic options to prevent VSMC proliferation both in nondiabetic and in diabetic states [13][45]. Therapeutic possibilities have also been studied by Chen et al., who analysed the effect of dasatinib, an orally bioavailable protein tyrosine kinase inhibitor, currently undergoing clinical trials in cancer patients [15][47]. Dasatinib is a potent PDGFR inhibitor at low nanomolar concentrations: comparison between dasatinib and imatinib showed that the former was able to decrease VSMC proliferation and inhibit PI3K/AKT at lower concentrations in A10 rat aortic smooth muscle cell lines [15][47]. To contrast VSMCs proliferation another interesting compound can be n-butylidenephthalide (BP), which was able to reduce PDGF-induced phenotypic switches both in vitro and in an AVF rat model, and it may contribute to vascular protection [16][48]. Yang et al., in fact, showed that BP can exert an anti-migrating effect, thus limiting the movement of VSMC from the media to the intima, and can also promote cells’ quiescent state by arresting cells in the G0/G1 cell cycle phase [16][48]. In rat fistula, BP can increase lumen size and decrease thrombus formation. Notably, these important effects are realized by the induction of AMPK and concomitant reduction of mTOR phosphorylation and signalling.
The PI3K/AKT/mTOR axis is also involved in extracellular matrix (ECM) formation: in fact, inflammation-released cytokines and growth factors can also influence extracellular matrix (ECM) degradation, which in turn helps VSMC infiltration and new matrix deposition. Notably, matrix metalloproteinase 9 (MMP-9) expression is upregulated in vascular inflammation and participates in vascular remodelling [12][17][16,49]. Recently, Shih et al. used MMP-9 knock-out mice with CDK and observed a decrease in inflammation, an increase in AVF lumen diameter, and attenuated neointima formation due to a concomitant reduction in collagen factor and smooth muscle cell proliferation, compared to wt CDK mice [12][16]. Since AKT and ERK phosphorylation increases in neointimal lesions in AVF, the authors showed that, by western blot experiments, the AKT and ERK phosphorylation increase is attenuated by MMP-9 knockout [12][16]. Importantly, PI3K/AKT/mTOR signalling repression can be related to a decrease in the inflammation, migration and proliferation of VSMC cells.
Since this AKT/mTOR involvement in cell growth and inflammation, inhibition of this signalling cascade by rapamycin treatment has been tested in diabetic patients and has shown efficacy in reducing neointimal hyperplasia by decreasing migration of smooth muscle cells and rapamycin. It is now currently used for patient therapy [18][50]. Dardik and colleagues extended involvement of AKT/mTOR signalling showing that it is upregulated in venous remodelling during both graft adaptation and AVF maturation [19][51]. Notably, amounts of Eph-B4 receptor as well as its ligand Ephrin-B2 increases in the vein wall during AVF maturation, and its signal is mediated through AKT activation [20][52]. Additional studies showed that rapamycin treatment caused a decrease in wall thickening due to diminished extracellular matrix deposition combined with a reduction in the proliferation of endothelial cells, muscle cells and macrophages [21][53]. Molecular investigation of this process demonstrated that rapamycin reduced activation of AKT/mTORC1 but not of mTORC2, and this was reflected in a reduction in the amounts of downstream targets of mTORC1, P70S6k and 4EPB1 [21][53], reinforcing the concept that inhibition of the AKT/mTORC1 axis with rapamycin reduces the pathologic venous remodelling that is associated with AVF failure.
To understand potential pathogenic steps in AVF failure, many studies have highlighted that remodelling of adventitia is caused also by the infiltration of myofibroblasts [22][54] accumulated in the adventitia. These myofibroblasts are responsible for fibrogenesis since they become a major source of collagen, producing an excess of the extracellular matrix at the base of the initiation and progression towards a pathological fibrotic vessel [23][55]. The exact source of the mesenchymal cells that produce collagen or other extracellular matrix components and become a central bulk of the mesenchymal cells participating in the fibrotic process is still not clear. Activated fibroblasts can originate from resident or vascular adventitial fibroblasts that pass from being quiescent to proliferative in response to signals received through the inflammation process [24][56]. They can also originate from epithelial cells that become myoblasts in response to signals such as transforming growth factor β (TGF-β) [25][57], and drive epithelial-to-mesenchymal transition (EMT) or endothelial-to-mesenchymal transition (EndoMT). In fact, EndoMt has emerged as a transdifferentiation biological process that induces endothelial differentiated cells to acquire a different (mesenchymal or myofibroblastic) phenotype and so able to produces mesenchymal proteins such as SMA, vimentin, and Type I collagen (Figure 1 3 and Figure 25). This transdifferentiation can be activated by the PDGF receptor along with a marked increase in the phosphorylation of Akt and ERK, two key kinases in PDGFRβ signalling [14][46]. Inhibition of this signalling cascade can reduce the presence of myofibroblasts that were the main cell type associated with the activation of p-PDGFRβ. The EndoMt process can be stimulated also by TGF-β released by lymphocytes and macrophages present due to the inflammatory state [23][55]. Notably, macrophages and lymphocytes play key and controversial roles in AVF inflammation and thus in AVF failure. High levels of inflammation (measured by c-reactive protein and fibrinogen) [26][58] are associated with AVF failure; however, depletion of macrophages and T cells also results in AVF failure. This controversial relationship can in part be explained by the different types and roles of immune cells in vascular wall formation and thus AVF patency and maturation (clearly reviewed in [27][9]).
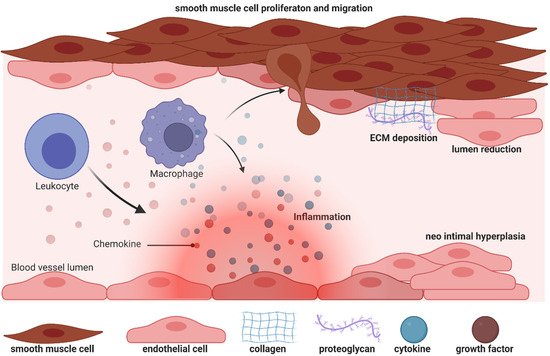
Figure 13.
Biology of efferent vein wall of AVF remodelling.
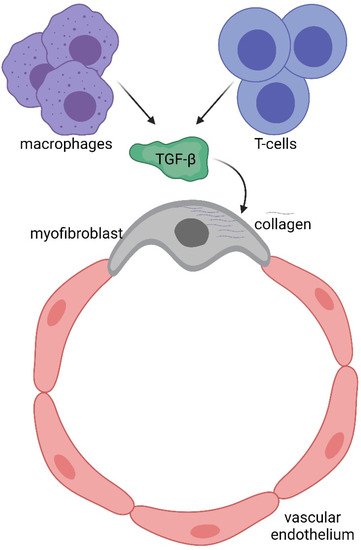
Figure 25.
Immune system involvement in endothelial-to-mesenchymal transition (EndoMT).
The immune system consists of a sophisticated network of cells which recognise and eliminate foreign threats to our body and provide immunological memory against external pathogens. Among the different immunological cell types, T cells provide adaptive immunity (acquired immune system), while macrophages (circulating monocytes that become resident next to the vessel wall) work as innate immunity. The two arms collaborate together during defensive processes such as inflammation. During AVF remodelling in the early phase, T cells contribute positively to augmented blood flow and outward remodelling, but unfortunately T regulatory (Treg), T helper2 (Th2) and M2 macrophages are related to late AVF failure [27][9]. Similarly (but also on the contrary), during the late phase, T helper1 (Th1) and M1 macrophages inhibit neointimal hyperplasia and thus enhance AVF patency, but unfortunately their presence is associated with primary AVF. Inhibition of AKT/mTORC1 signalling by sirolimus is now being used in clinical patients to help AF patency, probably by regulating Treg cells [28][59]. It is possible that in the future different inhibitors will be used to selectively limit specific cells of the immune system and improve clinical outcomes.