Extracellular vesicles (EV) are a very diverse group of cell-derived vesicles released by almost all kind of living cells. EV are involved in intercellular exchange, both nearby and systemically, since they induce signals and transmit their cargo (proteins, lipids, miRNAs) to other cells, which subsequently trigger a wide variety of biological responses in the target cells. However, cell surface receptor-induced EV release is limited to cells from the immune system, including T lymphocytes. T cell receptor activation of T lymphocytes induces secretion of EV containing T cell receptors for antigen and several bioactive molecules, including proapoptotic proteins. These EV are specific for antigen-bearing cells, which make them ideal candidates for a cell-free, EV-dependent cancer therapy. In this review we examine the generation of EV by T lymphocytes and CAR-T cells and some potential therapeutic approaches of these EV.
- exosomes
- T lymphocytes
- immune synapse
- secretory granules
- multivesicular bodies
- cytotoxic activity
- cell death
- CAR T lymphocytes
1. Introduction
1.1 Extracellular Vesicle Types
Extracellular Vesicle Types
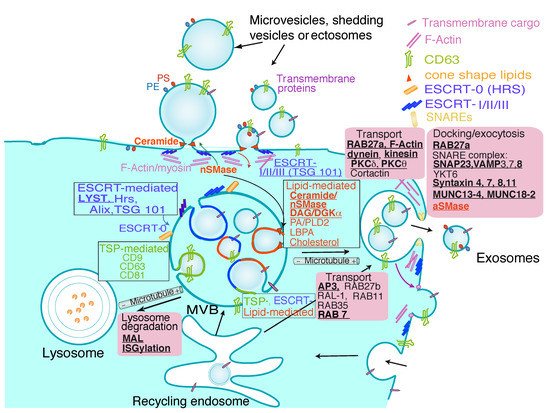
2. Extracellular Vesicles from T Lymphocytes
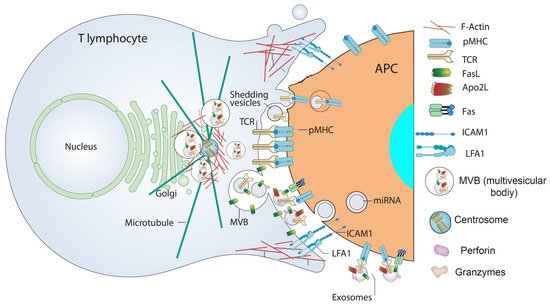
IS formation by T lymphocytes subsequent to TCR binding to antigen bound to MHC on the APC surface is a very dynamic, plastic and critical outcome involved in antigen-specific, cellular and humoral immune responses [48][49][93,94]. IS establishment integrates signals and combines molecular interactions leading to a proper and antigen-specific immune response [50][95]. There are two main groups of IS built by T lymphocytes leading to quite different, although essential, immune effector functions [48][50][51][93,95,96]. The interaction of helper T lymphocytes (Th), generally CD4+ cells, with MHC-II-bearing APC causes T lymphocyte activation (cytokine secretion, proliferation, etc.). In contrast, naïve CTL, usually CD8+ cells, recognize antigen-associated MHC-I on APC and become activated or “primed” to proliferate in the first phase, and kill target cells bearing the antigen in the second, effector phase. In the effector phase, primed CTL similarly form IS with target cells (virus-infected cells or tumor cells) leading to specific killing. Thus, the functional outcomes produced by the formation of an effective, mature IS include activation (naïve CTL and Th lymphocytes), killing (primed CTL), and functional anergy or apoptosis induction [52][97] once the effector phase is finished. IS formation induces the convergence of T lymphocyte secretion granules towards the MTOC and, almost simultaneously, MTOC polarization and secretion granules move towards the central supramolecular activation cluster (cSMAC) at the IS [49][53][94,98]. T lymphocyte secretion granules include cytokine-containing secretion granules in Th lymphocytes, cytolytic granules/secretory lysosomes in CTL, and MVB in Th lymphocytes and CTL. This dedicated mechanism appears to specifically endow the immune system with a superbly tuned tactic to enhance the efficiency of decisive secretory effector roles of T lymphocytes, while diminishing nonspecific, cytokine-controlled stimulation, target cell killing and activation induced cell death (AICD) of bystander cells [54][99]. CTL cytolytic granules are secretory lysosomes that have an MVB structure, and their degranulation causes ILV secretion as nanosize “extracellular vesicles” at the synaptic cleft made at the CTL-target cell interface, formerly described by Peters et al. [55][100]. Although CTL-secreted vesicles were not referred in this study as canonic exosomes, their creation and mode of exocytosis warrants this classification [10] (see above). Cytolytic granules contain perforin and granzymes that are located in a lumenal, electron dense core characterized by EM and also accumulate in the ILV [56][77]. In addition, it was shown that ILV and their derived exosomes contained, apart from the proapoptotic proteins perforin and granzymes, the exosome marker CD63 and molecules relevant for CTL-target cell interaction, such as TCR and CD8 [56][55][77,100], demonstrating that most of the cytotoxic factors exocytosed into the cleft between CTL and the target cell are membrane-enveloped or exosome-associated. However, release of perforin and granzymes in soluble form coming from the lumenal core cannot be excluded [56][57][77,101]. Perforin, which is inactive in the acidic environment of secretory lysosomes, is activated by neutral pH and Ca2+ at the synapse and polymerizes and forms a transmembrane pore that allows the entry of granzymes into the target cell; granzymes trigger caspase-dependent and independent cell death [44]. It was hypothesized that the presence of the TCR complex, CD8, and possibly other relevant molecules on these nanovesicles displaying their extracellular portions facing outwards, may ensure unidirectional delivery of lethal factors to target cells, since it was proposed that ILV released into the synaptic cleft bind specifically to the relevant antigen-MHC-I complex on the target cell membrane, and not to the CTL itself or to bystander cells [58][79]. This model would explain not only why a CTL does not kill itself, but also why bystander cells, which are in close proximity but do not bear the proper antigen, are not killed [58][79]. Subsequently, it was demonstrated that newly synthesized Fas ligand (FasL) is also stored in the limiting membrane of CTL secretory lysosomes and that polarized degranulation controls FasL delivery to the T cell surface, which is consistent with the role of a FasL-dependent pathway in CTL-mediated cytotoxicity [59][102]. In addition, it was shown that FasL can also be sorted from the MVB limiting membrane to ILV and hence to exosomes upon T lymphocyte activation, since T lymphocyte activation induced 100–200 nm “microvesicles” secretion including proapoptotic FasL and Apo2L [60][103] (Figure 2 and Figure 3). These microvesicles were subsequently characterized as canonic exosomes, since they arose from FasL+Apo2L+ ILV after MVB fusion with the plasma membrane [61][56]. Exosomal FasL and Apo2L, with the same topology as cell surface FasL and Apo2L, can bind to their respective death receptors on the surface of target cells, or effector T lymphocytes themselves, inducing caspase-dependent apoptosis [62][63][104,105] (Figure 3). Proapoptotic exosomes are thus involved in AICD of effector T lymphocytes, which constitutes an important suicide or fratricide mechanism participating in the downregulation of T cell-dependent immune responses [61][60][64][56,103,106]. Another major contribution was to demonstrate that inducible, polarized exosome secretion occurred at the IS formed by living Th lymphocytes and APC [18], as occurred in the IS formed by CTL [56][55][77,100] (Figure 3).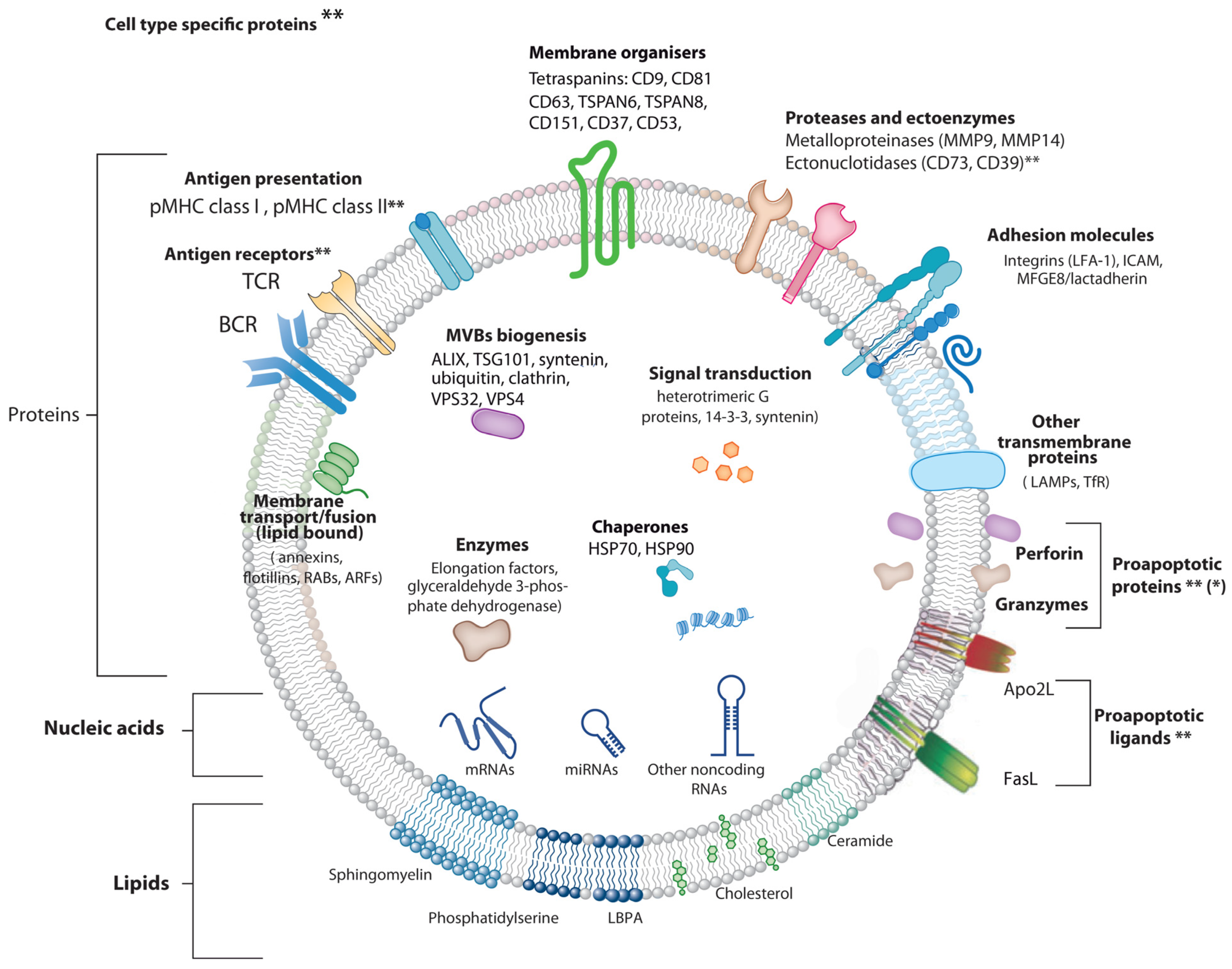
3. Chimeric Antigen Receptor (CAR) T Cells and CAR T Cell-Derived EV
3.1 Cancer Therapeutic Approaches
Cancer Therapeutic Approaches
| Target Molecule | EV-Producing Cell | EV Types | EV Phenotype | Anti-Tumor Mechanism | Target Cell | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EGFR, HER2 [81] | EGFR, HER2 [55] |
Human CAR T cells (?) | 1 | Exosomes | CAR | + | , CD3 | + | , CD63 | + | , perforin | + | , granzyme B | + | , CD45 | − | , CD28 | − | Perforin/ granzyme B | 2 | EGFR | + | , HER2 | + | human breast cancer cells |
| HER2 [84] | HER2 [138] |
Human CAR T cells CD4 | + | (46%) CD8 | + | (49%) | EV (small EV, probably exosomes plus larger EV) | 3 | CAR | + | , CD3 | + | , CD63 | +, | granzyme B | + | Granzyme B | 2 | HER2 | + | human breast cancer cells, ovarian cancer cells |
||||
| Mesothelin [83] | Mesothelin [80] |
Human CAR T cells CD4 | + | (58%) CD8 | + | (31%) | −Probably exosomes | 4 | CAR | + | , CD3 | + | , CD63 | + | , perforin | + | , granzyme B | + | Perforin/ granzyme B | 2 | Triple negative human breast cancer cells |
||||
| ++ | CD19 [85] | CD19 [139] |
Human CAR HEK293 cells | Probably exosomes | 4 | CAR | + | , CD63 | + | , CD81 | + | Indirect induction of proapoptotic genes in target cells |
CD19 | + | human B cell leukemia | ||||||||||
| Efficiency against solid tumors | +/− | ++ | CD19 [82] | CD19 [137] |
Human CAR HEK293 cells | Probably shedding vesicles | 4 | CAR | + | , annexin V binding (PS exposure) |
MYC Gene disruption mediated by CRISPR/Cas9 |
CD19 | + | human B cell leukemia cell lines | |||||||||||
| Immunosuppression by tumoral PD-L1 | + | − | Mesothelin CD19 [86] | Mesothelin CD19 [140] |
Human and mouse CAR T Cells (?) | 1 | EV | 4 | Unknown | 5 | Contain RN7SL1 |
Recruitment of endogenous anti-tumor immunity byRN7SL1 |
Mouse melanoma expressing human CD19 |
| Event | CAR T Cells | CAR T Cell-Derived EV | |
|---|---|---|---|
| Cytokine releasing syndrome | ++ | − | |
| Neurotoxicity | ++ | − | |
| Cross the blood barrier | |||
| Immunological memory | |||
| + | 1 | (?) | 2 |