Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 3 by Rita Xu.
Pancreatic cancer is one of the deadliest cancers worldwide, largely due to its aggressive development. Consequently, treatment options are often palliative, as only one-fifth of patients present with potentially curable tumors. The only available treatment with curative intent is surgery followed by adjuvant chemotherapy. However, even for patients that are eligible for surgery, the 5-year OS remains below 10%.
- PDAC
- TME
- cancer therapy
- pancreatic cancer
1. Introduction
Pancreatic cancer, in its most common form—pancreatic ductal adenocarcinoma (PDAC)—is one of the most lethal cancers worldwide. PDAC is the third leading cause of cancer death and is expected to become the first within the next ten years. With its five-year survival rate of 7%, PDAC is characterized by its aggressive nature and rapid metastasis formation. Moreover, the symptoms of PDAC, such as back pain, loss of appetite, weight loss and new-onset diabetes [1], are not specific and are often misinterpreted, leading to a late diagnosis [2]. In fact, less than 20% of PDAC patients present local and potentially curable tumors. While the cause of pancreatic cancer remains unknown, some factors such as smoking [3], chronic pancreatitis, which can be caused by excessive alcohol consumption [4][5], and age have been associated with an increased risk for the development of PDAC [6][7]. Furthermore, some pathogenic germline gene variants are known to increase susceptibility to PDAC, mainly occurring in DNA damage repair genes. The most observed variants in PDAC include BRCA1/2 (breast cancer gene 1/2) and ATM (ataxia telangiectasia mutated).
In most cases, pancreatic cancer originates in the ductal epithelium and develops from pre-malignant lesions. The best-characterized pre-malignant lesion and precursor of pancreatic cancer is called pancreatic intraepithelial neoplasia (PanIN) [8]. As cancer develops, the minimally dysplastic epithelium, called pancreatic intraepithelial neoplasia grades 1A and 1B, progresses to more severe dysplasia called pancreatic intraepithelial neoplasia grades 2 and 3 to finally reach the stage of an invasive carcinoma. In parallel, successive mutations accumulate, including the activation of the KRAS oncogene, the inactivation of the tumor-suppressor gene CDKN2A, and finally the inactivation of the tumor-suppressor gene TP53 and deleted in pancreatic cancer 4 (DPC4), also known as the SMAD family member 4 (SMAD4). The inactivation of CDKN2A results in the loss of the p16 protein, a regulator of the G1-S transition of the cell cycle, and the activation of TP53 allows cells to bypass DNA damage control checkpoints. The mutation of SMAD4 results in aberrant transforming growth factor β (TGF-β) signaling. Other less well characterized premalignant lesions of the pancreas have been described and characterized as intrapancreatic mucinous neoplasia (IPMN) and mucinous cystic neoplasia (MCN) [9]. The Genome Project showed that PDAC is a type of tumor with high inter-tumoral genetic heterogeneity [10]. This suggests that it might not be possible to find a single therapy for all PDAC patients. In fact, two major molecular subtypes of PDAC have recently been described, namely the classical and the basal-like subtypes. The classical subtype is characterized by a higher differentiation of the tumor, fibrosis, and inflammation, while the basal-like subtype shows a loss of differentiation and is associated with poor survival and a lack of response to existing chemotherapy regimens [11]. These findings support the fact that PDAC presents a high inter-patient variability and that treatment regimens may have to be adapted to the different tumor subtypes.
2. PDAC Standard-of-Care Treatments
To optimally treat PDAC patients, tumor staging is a crucial step. The staging of pancreatic cancer is based on the results of helical CT and tumor-node-metastasis classification according to the most recent edition of the ‘American Joint Committee on Cancer’ [12] (Table 1). The goal of CT imaging is to determine the position and size of the tumor, the involvement of veins and arteries, and the presence of metastasis. In brief, stages I and II comprise tumors that are limited to the pancreas and have minimal or no contact with major vessels. The difference between stage I and II is the size of the tumor (≤2 cm for I and >2 cm for II). Tumors of stage III are extended beyond the pancreas, involving the superior mesenteric vein, portal vein or splenic vein, but do not involve the celiac axis or the superior mesenteric artery. These three tumor stages can include regional lymph node metastasis. Stage IV tumors involve the superior mesenteric artery or celiac axis [13]. The tumor stage is a benchmark for assessing resectability. Stage I and II are considered resectable, stage II-III tumors are borderline resectable (BRPC) or locally advanced PDAC (LAPC), depending on whether a safe and complete resection and the reconstruction of the affected veins and arteries are possible. In general, the borderline resectable status indicates that the tumor is neither clearly resectable nor clearly unresectable but rather involves a greater likelihood of incomplete resection, including R1 resection and a positive margin, as part of upfront surgery [14]. Finally, stage IV tumors are metastatic PDAC and are considered non-resectable [15]. Precise tumor staging allows clinicians to provide the optimal treatment to the patients based on the advancement of the disease. Within the next sections, we will review the recommended treatments depending on the tumor stage.
Table 1. Overview of pancreatic cancer staging.
| AJCC Staging | I–II | II–III | II–III | IV |
|---|---|---|---|---|
| Clinical stage | Resectable | Borderline resectable | Locally advanced | Metastatic |
| Vascular involvement | No or <180° contact | <180° contact | >180° contact | N/A |
| Prevalence at diagnosis (~%) a | 10–15 | 30–35 | 30–35 | 50 |
| Treatment intent | Curative | Curative | Palliative | Palliative |
| 5-year survival rate (~%) a | 35–45 | 10–15 | 10–15 | <5 |
a Cancer Statistics, 2021; CA CANCER J CLIN 2021;71:7–33; doi: 10.3322/caac.21654.
2.1. Surgery
Today, surgery remains the only cure for patients with pancreatic cancer, but less than 20% of them are eligible for surgery. The main goal of surgery is to resect the entire tumor, in a way that no cancer cells can been seen microscopically at the primary tumor site. This successful resection is termed R0. On the other hand, a resection is called R1 when cancer cells remain microscopically visible at the primary tumor site. Unfortunately, a recent analysis concluded that the proportion of R1 resection exceeds 75%, even in specialized surgical centers [16]. The Whipple procedure, the resection of pancreatic head adenocarcinoma, consists of pancreatoduodenectomy, which includes the resection of the pancreatic head, duodenum, distal common bile, and sometimes gastric antrum, followed by pancreatoenterostomy, hepaticojejunostomy and gastrojejunostomy. This procedure is associated with a high morbidity rate of up to 45%. When the tumor is found in the tail or the body of the pancreas, the performed surgery is termed distal pancreatectomy, where the tail and possibly a part of the body of the pancreas is resected. As a safety measure due to proximity, the spleen is often removed as well. Finally, if the cancer has spread throughout the pancreas but is still resectable, a total pancreatectomy is performed, whereby the entire pancreas is removed, as well as the gallbladder, the spleen and parts of the stomach and the small intestine. This type of resection is the most drastic and is avoided as much as possible due to major side effects, such as diabetes and the inability to digest certain foods. Therefore, patients that have undergone total pancreatectomy are entirely dependent on insulin shots and must take pancreatic enzymes for life.
2.2. Neoadjuvant Chemotherapy
Neoadjuvant therapy can eradicate metastases that are not visible with imaging methods and in some cases, tumor shrinkage has been observed, which improves the resectability [17]. However, the benefit of neoadjuvant therapy compared to upfront surgery and adjuvant therapy for resectable PDAC is undergoing evaluation. This is addressed in the ongoing phase III trial (A021806) where perioperative FOLFIRINOX (Ca2+ folinate, 5-FU, irinotecan and oxaliplatin) is compared to adjuvant FOLFIRINOX for patients with resectable PDAC [18]. For BRPC tumors, surgery is often an option but is likely to result in R1 resection, which is an important negative prognostic factor for overall survival (OS) [19]. In these cases, neoadjuvant chemotherapy, such as FOLFIRINOX, may increase the rate of successful R0 resection and improve long-term survival [20]. Although neoadjuvant treatment is recommended by the National Comprehensive Cancer Network (NCCN), it has not yet become the standard-of-care treatment due to the lack of randomized studies and the added complexity to the multidisciplinary treatment planning. Neoadjuvant therapy requires a pretreatment biopsy and endoscopic stent placement in patients with biliary obstruction. However, a meta-analysis that included 13 trials demonstrated the down-staging of patients with BRPC or unresectable tumors after neoadjuvant FOLFIRINOX therapy, with an R0 resection rate of 40% [21]. Another meta-analysis highlighted the benefits of neoadjuvant therapy in resectable and borderline resectable pancreatic cancer, by comparing survival by intention to treat between patients undergoing upfront surgery and neoadjuvant chemotherapy. A total of 3484 patients were included in the analysis and approximately 50% of them received neoadjuvant treatment. Despite a lower resection rate (66% vs. 81.3%), patients who received neoadjuvant therapy survived longer compared to upfront surgery (18.8 vs. 14.8 months, respectively). The difference was even larger among patients who underwent resection (26.1 vs. 15 months) [22].
2.3. Adjuvant Chemotherapy for Resected Pancreatic Cancer
The randomized trial of the ESPAC-1 demonstrated that adjuvant fluorouracil (5-FU) chemotherapy has a beneficial impact on the OS of the patients after R0 and R1 resection (Table 2) [23]. The phase III CONKO-001 trial showed similar positive results after gemcitabine treatment, with prolonged disease-free survival (DFS) and OS, including in patients with R0- and R1-resected tumors (Table 2). Long-term follow up also showed an increased 10-year OS of 5% [24][25]. Finally, the phase III ESPAC-3 trial compared adjuvant therapy with either gemcitabine or 5-FU plus folinic acid and concluded that there was no significant difference between the two options [26] (Table 2). Therefore, both are currently considered standard treatments [15]. The benefit of adjuvant radiotherapy remains controversial. Therefore, treatment with systemic chemotherapy has become the standard of care after R0 and R1 resection. Recently, impressive progress has been achieved with FOLFIRINOX as adjuvant treatment for patients with resected PDAC. As shown in the PRODIGE 24/CCTG PA.6 trial, the median OS of the FOLFIRINOX group reached 53.5 months compared to 35.5 months in the gemcitabine group. A recent update of this trial included the 5-year patient follow up. The 5-year DFS rate is 26.1% for patients treated with FOLFIRINOX vs. 19.0% for gemcitabine, and the 5-year OS rate is 43.2% vs. 31.4%, respectively [27] (Table 2). Although FOLFIRINOX does not cure PDAC, these data demonstrate its positive impact on patient survival and are another step in the right direction. Therefore, FOLFIRINOX is recommended as adjuvant treatment for patients with excellent health status. For patients with poorer general health status, gemcitabine remains the preferred option.
Table 2. Summary of selected clinical trials.
| Trial | Therapy Type | Treatment Groups | Number of Patients | Median Survival in Months | Year of Publication |
|---|---|---|---|---|---|
| ESPAC-1 | Adjuvant | 5-FU vs. resection only | 188 | 19.7 vs. 14 | 2001 |
| CONKO-001 | Adjuvant | Gemcitabine vs. resection only | 368 | 13.4 vs. 6.7 | 2007 |
| ESPAC-3 | Adjuvant | 5-FU + folinic acid vs. gemcitabine | 1088 | 23 vs. 23.6 | 2010 |
| PRODIGE | First line for stage IV | FOLFIRINOX vs. gemcitabine | 342 | 11.1 vs. 6.8 | 2011 |
| PRODIGE-24 | Adjuvant | FOLFIRINOX vs. Gemcitabine | 493 | 54.4 vs. 35 | 2017 |
2.4. Treatment of Locally Advanced and Metastatic Disease
LAPC is inoperable and up to 80% of the patients will not have sufficient tumor response to neoadjuvant chemotherapy to become eligible for resection. The standard treatment aims to control the disease with chemotherapy, commonly FOLFIRINOX, nab-paclitaxel and/or gemcitabine [28], however, the OS of LAPC patients remains below one year [29]. Clinical trials have shown the equivalent response and efficacy of nab-paclitaxel and gemcitabine or FOLFIRINOX [30][31]. These are therefore considered as standard-of-care treatments and the use of either one is adapted based on the degree of adverse effects observed in each patient [32]. However, for approximately 20% of the patients who respond well to chemotherapy combinations, such as FOLFIRINOX or gemcitabine plus nab-paclitaxel, some studies reported promising results, namely a reduction in tumor size, converting some LAPC into resectable tumors [21].
Chemotherapy remains the standard treatment for metastatic pancreatic cancer. In fact, gemcitabine has been the standard of care for first-line treatment for the past 14 years, as a study has shown its clinical benefits in improving median progression-free survival (PFS). Unfortunately, even with gemcitabine treatment, the OS of patients with metastatic disease is only between five and six months with a response rate of 5.4% [33]. Various combinations of gemcitabine-based treatments with either cytotoxic or molecularly targeted agents have been tested and showed only a slight increase in the OS, from a few weeks to a few months [30][33][34][35][36]. Similarly, several meta-analysis of randomized controlled trials have shown that combining gemcitabine with fluoropyrimidine or platinum compounds moderately improves OS in patients with advanced PDAC [37][38][39]. In the PRODIGE trial of 2011, an important breakthrough for patients with metastatic PDAC was achieved with FOLFIRINOX treatment. The OS of patients treated with FOLFIRINOX increased up to 11.1 months compared to the 6.8 months observed with gemcitabine alone [35] (Table 2). Since this trial, FOLFIRINOX has become the standard of care for patients with metastatic PDAC. This major discovery then led to the use of FOLFIRINOX as adjuvant and neoadjuvant therapy. Recently, Shelemey et al. reported the case of a 59-year-old woman with adenocarcinoma of the pancreatic tail and innumerable liver metastases who received FOLFIRINOX chemotherapy. Subsequent CT scans showed shrinkage of the pancreatic mass, as well as the liver metastases. Her cancer antigen 19-9 (CA 19-9) normalized after 11 months. Oxaliplatin was then discontinued due to peripheral neuropathy, but she still completed 37 cycles of FOLFIRI (Ca2+ folinate, 5-FU, irinotecan). During these cycles, the pancreatic mass disappeared, and the liver metastases decreased in size and remained as scar tissue. After 5.5 years, an MRI of her abdomen showed no recurrence of pancreatic mass, and two residual liver lesions, defined as scar tissue, remained stable [40]. This case of complete response to FOLFIRINOX/FOLFIRI chemotherapy is encouraging and argues for its use as standard therapy.
3. PDAC TME, Immunotherapies and Therapy Resistance: What to Target Next?
After reviewing the currently available treatments based on tumor stage, rwesearchers now discuss the major features of PDAC and its TME. In the next section, theywe will consider possible causes of treatment resistance and how to identify potential new therapeutic targets based on what is known about the TME in PDAC (Figure 1).
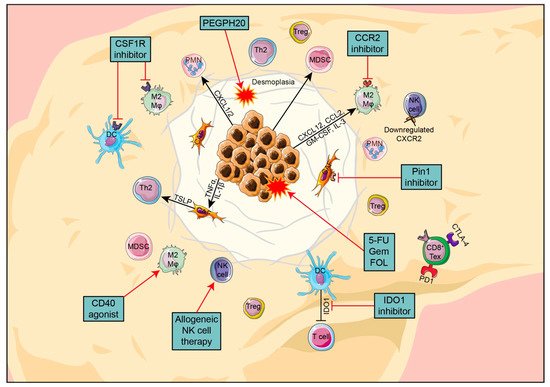
Figure 1. Overview of the key components in PDAC TME, their interactions and promising new targets.
3.1. Pancreatic Cancer Cells
During the development of PDAC, pancreatic cancer cells acquire several features that enable them to evade the immune system. First of all, cancer cells are able to downregulate their expression of major histocompatibility class I (MHC-I) molecules, making them less well recognized by effector T cells [41][42]. Second, PDAC cells can attract various immunosuppressive cells that further contribute to the low immunogenicity of the tumor. For example, PDAC cells use the CCL2/CCR2 axis to recruit tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) [43], and the CCL5/CCR5 axis to recruit regulatory T cells (Tregs) [44]. Third, pancreatic cancer cells can reduce effector cell function within the TME by the immune checkpoint molecule programmed death-ligand 1 (PD-L1), which is expressed in approximately 13% of PDAC patients. PD-L1 binds to PD-1 on the T cells and induces their anergy and apoptosis, contributing to immune evasion [45]. Furthermore, PDAC cells produce indoleamine 2,3-dioxygenase (IDO), which catalyzes tryptophan degradation. As tryptophan is required for T cell survival and activation, its degradation also leads to T cell apoptosis and anergy [46]. In addition, it has been shown that IDO enhances the recruitment of Tregs and tryptophan starvation induces their development in the TME [47][48]. Together, these mechanisms allow pancreatic cancer cells to develop and proliferate while being protected from an immune response.
3.2. PDAC Stroma
Upon PDAC establishment, one major characteristic is a desmoplastic reaction which is the formation of dense stroma [49]. This stroma is composed of high-density fibrotic tissue and pancreatic stellate cells (PSCs), which are the cancer-associated fibroblasts (CAFs) specific to PDAC and the main feature of PDAC, accounting for nearly 90% of the tumor mass. The formation of a dense stroma is the result of PSCs or myofibroblasts activation by growth factors such as TGF-β1, platelet-derived growth factor (PDGF) and fibroblast growth factor. Activated PSCs secrete collagen and other components of the extracellular matrix which play a role in poor vascularization, which is characteristic of PDAC [50]. However, the role of stromal cells in pancreatic cancer progression seems controversial. In some studies, the presence of stroma has been shown to promote immunosuppression and fibrosis [51][52] and support cancer progression by attenuating antitumor effector mechanisms. The stroma itself increases the number of immunosuppressive cells and inhibits cytotoxic CD8+ T cells [53]. In these studies, it seems that the stroma does not only constitute a physical barrier, but also forms a compartment involved in the process of tumor formation, progression, invasion and metastasis [49]. It has been shown that stromal cells express proteins associated with poor prognosis and treatment resistance, such as PDGF receptor, vascular endothelial growth factor (VEGF) and secreted protein, acidic and rich in cysteine (SPARC) [54][55]. However, Özdemir et al. conducted a preclinical study in which CAFs were depleted and surprisingly, they observed that collagen was reduced and matrix was reorganized, angiogenesis was decreased, and hypoxia was enhanced. In addition, the number of cancer stem cells and the frequency of Tregs increased, leading to a poor prognosis [56]. Recently, much progress has been made in deciphering the controversial role of CAFs, which is now known to be due to several contrasting functions. Distinct CAFs populations have been identified that differ in their gene expression and secretome profiles. Myofibroblastic CAFs (myCAFs) are the closest to cancer cells with properties of activated fibroblasts. On the other hand, inflammatory-CAF (iCAFs) are in a more distal location from the tumor and are characterized by the expression of inflammatory mediators, such as IL-1, IL-21, and CXCL1-3 [57][58].
3.3. Treatments Targeting PDAC Stroma
The desmoplastic stroma of PDAC not only constitutes a large portion of the tumor mass, but also plays a key role in immunosuppression and acts as a barrier preventing effective therapy. However, treatments targeting the stroma have revealed controversial results, and it is not clear whether the modulation of the stroma is beneficial in PDAC [56][59]. Interestingly, the most clinically advanced stromal modulator, hyaluronidase PEGPH20, has been tested in a phase II trial and has been shown to degrade stromal proteins, improve vascular perfusion, and prolong PFS in combination with standard chemotherapy [60]. Unfortunately, in a Phase III study, PEGPH20 in combination with chemotherapy failed to improve the clinical outcome of PDAC patients [61]. Another stromal modulator and regulator of TME fibrosis and immunosuppression, focal adhesion kinase (FAK), is associated with decreased T cell infiltration when highly expressed in human PDAC. In preclinical studies, the inhibition of FAK enhanced the response to chemotherapy and checkpoint blockade, while reducing the infiltration of MDSCs into the TME [62]. Further studies are required to understand the mechanisms and benefits of targeting the desmoplastic stroma in PDAC.
3.4. The Immune Compartment in PDAC
As previously discussed, the TME in PDAC is mainly composed of cancer cell nests and stroma. The stroma itself consists of stromal matrix and immune cells. Immune cells account for up to 50% of the total cell number in PDAC; however, only a small subset of these are tumoricidal cells, whilst the rest are tumor-promoting and immunosuppressive cells [63]. One of the reasons is the release of immunosuppressive cytokines such as IL-10 and TGF-β and the recruitment of immunosuppressive cells during tumorigenesis [64][65]. IL-10 and TGF-β induce Tregs, which also produce IL-10 and TGF-β, creating a positive feedback loop that contributes to the inhibition of effector T cells and the maintenance of immunosuppression [66][67][68]. The immune compartment of PDAC is heterogeneous, the main immune cell types being dendritic cells (DCs), macrophages, neutrophils, MDSCs, natural killer cells (NKs), and effector T cells [69]. In the next section, reswearchers will elaborate on the main features of these various cell types, their role in PDAC, and their potential as therapeutic targets.
3.4.1. Dendritic Cells
In PDAC, the immunogenic and pro-inflammatory functions of myeloid cells are largely impaired. DCs have been shown to infiltrate PDAC lesions and their number increases with disease progression from PanIN to PDAC. However, the expression of DC maturation markers, such as MHC class II and the costimulatory molecules CD86 and CD40, is reduced by the action of Tregs, which directly affects CD8+ T cell activation and the expansion of tumor-infiltrating DCs [70]. DC function is further impaired by PDAC epithelial cells through the secretion of DC-suppressive cytokines such as IL-10, which downregulate MHC class I and CD40 expression, maintaining DCs in an immature stage [71][72].
3.4.2. Macrophages
In steady-state conditions or upon inflammation, most tissue macrophages originate from bone marrow-derived monocytes in the blood circulation. The remaining portion of macrophages are termed specialized tissue-resident, such as alveolar macrophages in the lungs, microglia in the brain, or Kupffer cells in the liver, and are not derived from blood monocytes [73]. When a tumor develops, it can recruit and induce macrophages in the TME with various chemokines and cytokines, such as CXCL12, CCL2, GM-CSF, colony-stimulating factor 1 (CSF-1), and IL-3. These special macrophages are then called tumor-associated macrophages (TAMs) [74][75]. A recent study showed that certain TAMs may also originate from tissue-resident macrophages, representing a functionally distinct subpopulation [76]. Although they form a continuous spectrum, TAM subpopulations are mainly divided into two opposite polarization states, namely M1 and M2. Typically, M1 macrophages are characterized by the secretion of pro-inflammatory cytokines and a tumoricidal function, while conversely, M2 macrophages secrete anti-inflammatory signals promoting tumor progression [77]. In PDAC, the majority of TAMs display an M2-like phenotype, characterized by the expression of surface markers CD163 and CD206, and the cytokines IL-10 and TGF-β [78]. They are primarily located at the invasive front of the tumor [63][79]. Macrophage infiltration begins early and persists throughout cancer progression [63]. TAM infiltration is correlated with perineural invasion [80], angiogenesis, lymph node metastasis [79][80][81], cancer cell epithelial–mesenchymal transition and extravasation [78]. Therefore, macrophage depletion reduces lung and liver metastasis in an orthotopic mouse model of PDAC [43]. Thus, TAMs appear to be involved in both regulating PDAC invasion and metastasis and are therefore correlated with worse OS. Finally, TAMs are partially responsible for the impaired efficacy of chemotherapy in PDAC. It has been shown that they are regulating the function of cytidine deaminase (CDA), which is a key metabolizer of gemcitabine, thereby contributing to gemcitabine-based chemotherapy resistance [82].
References
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26.
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA Cancer J. Clin. 2013, 63, 318–348.
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049.
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261.
- Genkinger, J.M.; Spiegelman, D.; Anderson, K.E.; Bergkvist, L.; Bernstein, L.; van den Brandt, P.A.; English, D.R.; Freudenheim, J.L.; Fuchs, C.S.; Giles, G.G.; et al. Alcohol Intake and Pancreatic Cancer Risk: A Pooled Analysis of Fourteen Cohort Studies. Cancer Epidemiol. Prev. Biomark. 2009, 18, 765–776.
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057.
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer 2020, 126, 2225–2249.
- Hruban, R.H.; Maitra, A.; Goggins, M. Pancreatic Intraepithelial Neoplasia. Definitions 2020, 28, 306–316.
- Takaori, K. Current understanding of precursors to pancreatic cancer. J. Hepatobiliary Pancreat. Surg. 2007, 14, 217–223.
- Biankin, A.V.; Maitra, A. Subtyping Pancreatic Cancer. Cancer Cell 2015, 28, 411–413.
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178.
- Edge, S.B.; Byrd, D.R.; Carducci, M.A.; Compton, C.C.; Fritz, A.G.; Greene, F.L. AJCC Cancer Staging Manual; Springer: New York, NY, USA, 2010; Volume 649.
- Bilimoria, K.Y.; Bentrem, D.J.; Ko, C.Y.; Ritchey, J.; Stewart, A.K.; Winchester, D.P.; Talamonti, M.S. Validation of the 6th edition AJCC pancreatic cancer staging system: Report from the National Cancer Database. Cancer 2007, 110, 738–744.
- Lopez, N.E.; Prendergast, C.; Lowy, A.M. Borderline resectable pancreatic cancer: Definitions and management. World J. Gastroenterol. 2014, 20, 10740–10751.
- National Comprehensive Cancer Network. Practice Guidelines in Oncology for Pancreatic Adenocarcinoma; Version 2; National Comprehensive Cancer Network: Jenkintown, PA, USA, 2021.
- Esposito, I.; Kleeff, J.J.; Bergmann, F.; Reiser, C.; Herpel, E.; Friess, H.; Schirmacher, P.; Büchlerbüchler, M.W. Most Pancreatic Cancer Resections are R1 Resections. Ann. Surg. Oncol. 2008, 15, 1651–1660.
- Ferrone, C.R.; Marchegiani, G.; Hong, T.S.; Ryan, D.P.; Deshpande, V.; McDonnell, E.I.; Sabbatino, F.; Santos, D.D.; Allen, J.N.; Blaszkowsky, L.S.; et al. Radiological and surgical implications of neoadjuvant treatment with FOLFIRINOX for locally advanced and borderline resectable pancreatic cancer. Ann. Surg. 2015, 261, 12–17.
- Alliance for Clinical Trials in Oncology. Testing the Use of the Usual Chemotherapy before and after Surgery for Removable Pancreatic Cancer; Alliance for Clinical Trials in Oncology: Boston, MA, USA, 2020.
- Fatima, J. Pancreatoduodenectomy for Ductal Adenocarcinoma. Arch. Surg. 2010, 145, 167.
- Delpero, J.R.; Boher, J.M.; Sauvanet, A.; Le Treut, Y.P.; Sa-Cunha, A.; Mabrut, J.Y.; Chiche, L.; Turrini, O.; Bachellier, P.; Paye, F. Pancreatic Adenocarcinoma with Venous Involvement: Is Up-Front Synchronous Portal-Superior Mesenteric Vein Resection Still Justified? A Survey of the Association Française de Chirurgie. Ann. Surg. Oncol. 2015, 22, 1874–1883.
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Lonati, V.; Aitini, E.; Barni, S. FOLFIRINOX-based neoadjuvant therapy in borderline resectable or unresectable pancreatic cancer: A meta-analytical review of published studies. Pancreas 2015, 44, 515–521.
- Versteijne, E.; Vogel, J.A.; Besselink, M.G.; Busch, O.R.C.; Wilmink, J.W.; Daams, J.G.; van Eijck, C.H.J.; Groot Koerkamp, B.; Rasch, C.R.N.; van Tienhoven, G.; et al. Meta-analysis comparing upfront surgery with neoadjuvant treatment in patients with resectable or borderline resectable pancreatic cancer. Br. J. Surg. 2018, 105, 946–958.
- Neoptolemos, J.P.; Stocken, D.D.; Friess, H.; Bassi, C.; Dunn, J.A.; Hickey, H.; Beger, H.; Fernandez-Cruz, L.; Dervenis, C.; Lacaine, F.; et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med. 2004, 350, 1200–1210.
- Oettle, H.; Post, S.; Neuhaus, P. Vs Observation in Patients Undergoing Curative-Intent Resection of Pancreatic Cancer. JAMA 2007, 297, 267–277.
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA J. Am. Med. Assoc. 2013, 310, 1473–1481.
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C. Adjuvant Chemotherapy with Fluorouracil Plus Folinic Acid vs. Gemcitabine Following Pancreatic Cancer Resection. JAMA J. Am. Med. Assoc. 2010, 304, 1073–1081.
- Conroy, T.; Hammel, P.; Turpin, A.; Belletier, C.; Wei, A.; Mitry, E.; Lopez, A.; Francois, E.; Artru, P.; Biagi, J.; et al. LBA57 Unicancer PRODIGE 24/CCTG PA6 trial: Updated results of a multicenter international randomized phase III trial of adjuvant mFOLFIRINOX (mFFX) versus gemcitabine (gem) in patients (pts) with resected pancreatic ductal adenocarcinomas (PDAC). Ann. Oncol. 2021, 32, S1334.
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B.; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M.; et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 439–457.
- Loehrer Sr, P.J.; Feng, Y.; Cardenes, H.; Wagner, L.; Brell, J.M.; Cella, D.; Flynn, P.; Ramanathan, R.K.; Crane, C.H.; Alberts, S.R.; et al. Gemcitabine Alone Versus Gemcitabine Plus Radiotherapy in Patients with Locally Advanced Pancreatic Cancer: An Eastern Cooperative Oncology Group Trial. J. Clin. Oncol. 2011, 29, 4105–4112.
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Wee Ma, W.; Saleh, N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 18, 1691–1703.
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel Plus Gemcitabine for Metastatic Pancreatic Cancer: Long-Term Survival From a Phase III Trial. JNCI J. Natl. Cancer Inst. 2015, 107, dju413.
- Cho, I.R.; Kang, H.; Jo, J.H.; Lee, H.S.; Chung, M.J.; Park, J.Y.; Park, S.W.; Song, S.Y.; An, C.; Park, M.-S.; et al. FOLFIRINOX vs gemcitabine/nab-paclitaxel for treatment of metastatic pancreatic cancer: Single-center cohort study. World J. Gastrointest. Oncol. 2020, 12, 182–194.
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413.
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966.
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825.
- Singhal, M.K.; Kapoor, A.; Bagri, P.K.; Narayan, S.; Singh, D.; Nirban, R.K.; Singh, G.; Maharia, S.; Kumari, P.; Jakhar, S.L.; et al. A Phase III Trial Comparing Folfirinox Versus Gemcitabine for Metastatic Pancreatic Cancer. Ann. Oncol. 2014, 25, iv210.
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2009, 27, 5513–5518.
- Li, Q.; Yan, H.; Liu, W.; Zhen, H.; Yang, Y.; Cao, B. Efficacy and Safety of Gemcitabine-Fluorouracil Combination Therapy in the Management of Advanced Pancreatic Cancer: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2014, 9, e104346.
- Heinemann, V.; Boeck, S.; Hinke, A.; Labianca, R.; Louvet, C. Meta-analysis of randomized trials: Evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer 2008, 8, 82.
- Shelemey, P.T.; Amaro, C.P.; Ng, D.; Falck, V.; Tam, V.C. Metastatic pancreatic cancer with complete response to FOLFIRINOX treatment. BMJ Case Rep. 2021, 14, e238395.
- Ryschich, E.; Nötzel, T.; Hinz, U.; Autschbach, F.; Ferguson, J.; Simon, I.; Weitz, J.; Fröhlich, B.; Klar, E.; Büchler, M.W.; et al. Control of T-Cell-Mediated Immune Response by HLA Class I in Human Pancreatic Carcinoma. Clin. Cancer Res. 2005, 11, 498–504.
- Pandha, H.; Rigg, A.; John, J.; Lemoine, N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin. Exp. Immunol. 2007, 148, 127–135.
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 2013, 19, 3404–3415.
- Tan, M.C.B.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.-S.; Linehan, D.C. Disruption of CCR5-Dependent Homing of Regulatory T Cells Inhibits Tumor Growth in a Murine Model of Pancreatic Cancer. J. Immunol. 2009, 182, 1746–1755.
- Basso, D.; Fogar, P.; Falconi, M.; Fadi, E.; Sperti, C.; Frasson, C.; Greco, E.; Tamburrino, D.; Teolato, S.; Moz, S.; et al. Pancreatic Tumors and Immature Immunosuppressive Myeloid Cells in Blood and Spleen: Role of Inhibitory Co-Stimulatory Molecules PDL1 and CTLA4. An In Vivo and In Vitro Study. PLoS ONE 2013, 8, e54824.
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274.
- Witkiewicz, A.; Williams, T.K.; Cozzitorto, J.; Durkan, B.; Showalter, S.L.; Yeo, C.J.; Brody, J.R. Expression of Indoleamine 2,3-Dioxygenase in Metastatic Pancreatic Ductal Adenocarcinoma Recruits Regulatory T Cells to Avoid Immune Detection. J. Am. Coll. Surg. 2008, 206, 849–854.
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761.
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197.
- Masamune, A.; Shimosegawa, T. Signal transduction in pancreatic stellate cells. J. Gastroenterol. 2009, 44, 249–260.
- Neesse, A.; Algül, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484.
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276.
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926.
- Mukherjee, P.; Basu, G.D.; Tinder, T.L.; Subramani, D.B.; Bradley, J.M.; Arefayene, M.; Skaar, T.; De Petris, G. Progression of Pancreatic Adenocarcinoma Is Significantly Impeded with a Combination of Vaccine and COX-2 Inhibition. J. Immunol. 2009, 182, 216–224.
- Infante, J.R.; Matsubayashi, H.; Sato, N.; Tonascia, J.; Klein, A.P.; Riall, T.A.; Yeo, C.; Iacobuzio-Donahue, C.; Goggins, M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol. 2007, 25, 319–325.
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734.
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301.
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747.
- Hingorani, S.R.; Bullock, A.J.; Seery, T.E.; Zheng, L.; Sigal, D.; Ritch, P.S.; Braiteh, F.S.; Zalupski, M.; Bahary, N.; Harris, W.P.; et al. Randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine (PAG) vs AG in patients (Pts) with untreated, metastatic pancreatic ductal adenocarcinoma (mPDA). J. Clin. Oncol. 2017, 35, 4008.
- Tempero, M.A.; Van Cutsem, E.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. HALO 109-301: A randomized, double-blind, placebo-controlled, phase 3 study of pegvorhyaluronidase alfa (PEGPH20) + nab-paclitaxel/gemcitabine (AG) in patients (pts) with previously untreated hyaluronan (HA)-high metastatic pancreatic ductal adenocarcinom. J. Clin. Oncol. 2020, 38, 638.
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860.
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527.
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676.
- Tanţău, A.; Leucuţa, D.-C.; Tanţău, M.; Boţan, E.; Zaharie, R.; Mândruţiu, A.; Tomuleasa, I.-C. Inflammation, Tumoral Markers and Interleukin-17, -10, and -6 Profiles in Pancreatic Adenocarcinoma and Chronic Pancreatitis. Dig. Dis. Sci. 2020, 66, 3427–3438.
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, 102655.
- Saka, D.; Gökalp, M.; Piyade, B.; Cevik, N.C.; Arik Sever, E.; Unutmaz, D.; Ceyhan, G.O.; Demir, I.E.; Asimgil, H. Mechanisms of T-Cell Exhaustion in Pancreatic Cancer. Cancers 2020, 12, 2274.
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163.
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133.
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571.
- Bellone, G.; Carbone, A.; Smirne, C.; Scirelli, T.; Buffolino, A.; Novarino, A.; Stacchini, A.; Bertetto, O.; Palestro, G.; Sorio, C.; et al. Cooperative Induction of a Tolerogenic Dendritic Cell Phenotype by Cytokines Secreted by Pancreatic Carcinoma Cells. J. Immunol. 2006, 177, 3448–3460.
- Bharadwaj, U.; Li, M.; Zhang, R.; Chen, C.; Yao, Q. Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res. 2007, 67, 5479–5488.
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188.
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964.
- Santoni, M.; Bracarda, S.; Nabissi, M.; Massari, F.; Conti, A.; Bria, E.; Tortora, G.; Santoni, G.; Cascinu, S. CXC and CC Chemokines as Angiogenic Modulators in Nonhaematological Tumors. Biomed Res. Int. 2014, 2014, 768758.
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896.
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1191731.
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219.
- Zeng, L.; Guo, Y.; Liang, J.; Chen, S.; Peng, P.; Zhang, Q.; Su, H.; Chen, Y.; Huang, K. Perineural invasion and TAMs in pancreatic ductal adenocarcinomas: Review of the original pathology reports using immunohistochemical enhancement and relationships with clinicopathological features. J. Cancer 2014, 5, 754–760.
- Kurahara, H.; Takao, S.; Maemura, K.; Mataki, Y.; Kuwahata, T.; Maeda, K.; Sakoda, M.; Iino, S.; Ishigami, S.; Ueno, S.; et al. M2-Polarized tumor-associated macrophage infiltration of regional lymph nodes is associated with nodal lymphangiogenesis and occult nodal involvement in pn0 pancreatic cancer. Pancreas 2013, 42, 155–159.
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819.
More